
Neuroblastooma lapsilla: syyt, diagnoosi, hoito
Viimeksi tarkistettu: 23.04.2024

Kaikki iLive-sisältö tarkistetaan lääketieteellisesti tai se tarkistetaan tosiasiallisen tarkkuuden varmistamiseksi.
Meillä on tiukat hankintaohjeet ja vain linkki hyvämaineisiin mediasivustoihin, akateemisiin tutkimuslaitoksiin ja mahdollisuuksien mukaan lääketieteellisesti vertaisarvioituihin tutkimuksiin. Huomaa, että suluissa ([1], [2] jne.) Olevat numerot ovat napsautettavia linkkejä näihin tutkimuksiin.
Jos sinusta tuntuu, että jokin sisältö on virheellinen, vanhentunut tai muuten kyseenalainen, valitse se ja paina Ctrl + Enter.
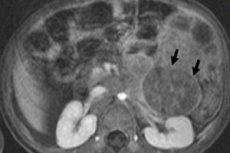
Lasten onkologiassa yksi yleisimmistä ekstrakraniaalisista kasvaimista on lasten neuroblastooma, joka viittaa pahanlaatuisiin alkion kasvaimiin hermoharjan neuroblasteista eli sympaattisen hermoston ituista (epäkypsistä) hermosoluista.
Epidemiologia
Kansainvälisen neuroblastoomariskiryhmän (INRG) tilastojen mukaan neuroblastooma muodostaa noin 8 % kaikista lasten syövistä maailmanlaajuisesti ja on levinneisyydessä kolmannella sijalla leukemian ja aivokasvainten jälkeen.
Muiden mukaan neuroblastooma aiheuttaa noin 28 % kaikista imeväisten syövistä. Yli kolmanneksessa tapauksista neuroblastooma diagnosoidaan alle vuoden ikäisillä lapsilla; Diagnoosin mediaani-ikä on 19-22 kuukautta. Yli 90 % diagnosoiduista tapauksista esiintyy 2–5-vuotiailla lapsilla (enimmäkseen poikia); ilmaantuvuushuippu on 2-3 vuoden iässä, ja tapaukset yli viisivuotiailla lapsilla ovat alle 10 %.
Syyt neuroblastooma
Tutkiessaan neuroblastooman syitä tutkijat päättelivät, että tämä kasvain lapsilla johtuu satunnaisista geneettisistä mutaatioista alkion synnyn tai varhaisen postnataalisen kehityksen aikana. Mutta mikä aiheuttaa nämä geenimuutokset, ei tiedetä, koska teratogeenisten ympäristötekijöiden vaikutusta ei ole tunnistettu.
Tällaisia kasvaimia voi esiintyä missä tahansa, mukaan lukien välikarsina, kaula, vatsa, lisämunuaiset, munuaiset, selkäranka, lantio.
Harvinaisissa tapauksissa imeväisten neuroblastooma voi johtua perinnöllisestä mutaatiosta. Erityisesti mutaatio CD246-membraaniproteiinigeenissä kromosomissa 2, ALK-tyrosiinikinaasientsyymissä, joka tarjoaa solujen välistä viestintää ja jolla on tärkeä rooli hermoston toiminnassa; PHOX2B-proteiinigeenissä (kromosomissa 4), joka osallistuu hermosolujen kypsymiseen.
Neuroblastooma voi liittyä myös tyypin 1 neurofibromatoosiin lapsilla, Beckwith-Wiedemannin oireyhtymään ja hyperinsulineemiseen hypoglykemiaan (haiman nesidioblastoosi).
Riskitekijät
Tähän mennessä perinnöllisyys on tunnustettu lasten neuroblastooman kehittymisen riskitekijöiksi - tämän kasvaimen esiintyminen suvussa sekä synnynnäiset poikkeavuudet, jotka liittyvät geenimutaatioihin sikiön kehityksen aikana. Tämä pätee erityisesti tapauksissa, joissa eri elimiin kehittyy useita kasvaimia.
Tutkijat eivät ole tunnistaneet mitään eksogeenisiä tekijöitä, jotka lisäävät tämän kasvaimen riskiä.
Synnyssä
Neuroblastoomien kehittymismekanismi johtuu hermoharjasolujen - kahdenvälisten solulinjojen - heikentymisestä ja kypsymisestä, jotka muodostuvat hermoputken reunoja pitkin ihmisalkion ektodermaalisesta itukerroksesta. Nämä solut vaeltavat (liikkuvat) ja erilaistuvat useiksi solutyypeiksi: sensorisiksi ja autonomisiksi hermosoluiksi, neuroendokriinisiksi ja lisämunuaisen ydinsoluiksi, kallonrusto- ja luusoluiksi sekä pigmenttisoluiksi.
Neuroblastoomassa siirtyneet neuroblastit eivät kypsy, vaan jatkavat kasvuaan ja jakautumistaan muodostaen kasvaimen. Ja sen muodostumisen patogeneesi liittyy seuraaviin geenimutaatioihin:
- osan kromosomisekvenssin tai RBTN1-proteiinia koodaavan LMO1-geenin segmenttien kaksinkertaistuminen kromosomissa 11 alkion hermoharjan soluissa;
- kopioluvun muutoksella NBPF10-geenissä kromosomissa 1q21.1, joka koodaa DUF1220-proteiinia, joka säätelee ihmisen hermokantasolujen lisääntymistä. Nämä häiriöt johtavat joko tämän kromosomin kaksinkertaistumiseen tai sen deleetioon - DNA:n osan puuttumiseen;
- kasvaimen suppressorigeenin ATRX muutoksilla (kromosomissa Xq21.1);
- kromosomissa 2 on lisäkopioita (amplifikaatiota) N-Myc-transkriptiotekijägeenistä, joka koodaa yhtä transkriptiotekijöistä (DNA:ta sitova proteiini), joka säätelee muiden geenien aktiivisuutta ja ohjaa kantasolujen lisääntymistä proteiinien muodostuminen sikiön kudosten ja elinten muodostamiseksi. Tämän geenin monistuminen muuttaa sen onkogeeniksi, mikä aiheuttaa solusyklin rikkomisen, lisää solujen lisääntymistä ja kasvainten muodostumista.
Oireet neuroblastooma
Ensimmäiset neuroblastooman merkit ovat epäspesifisiä ja voivat ilmetä ruokahaluttomuuden (ja painon laskun), ruokinnan aikaisen väsymyksen, kuumeen ja nivelkipujen muodossa.
Kliiniset oireet riippuvat primaarisen kasvaimen sijainnista ja etäpesäkkeiden esiintymisestä (näitä esiintyy 60–73 %:ssa tapauksista).
Hyvin usein primaarinen neuroblastooma sijoittuu lisämunuaisen ytimeen, jolla on samanlainen alkuperä hermosolujen kanssa. Vuoden iässä lasten lisämunuaisen neuroblastooma diagnosoidaan 35–40 prosentissa tapauksista. Sen oireita ovat vatsakipu, kuume, laihtuminen, luukipu, anemia tai samanaikainen Pepperin oireyhtymä: diffuusi maksavaurio, johon liittyy vaikea hepatomegalia ja hengitysvaikeusoireyhtymä.
Retroperitoneaalinen neuroblastooma tai retroperitoneaalinen neuroblastooma lapsilla alkaa kasvaessaan rasittaa virtsarakkoa tai suolia, mikä voi aiheuttaa virtsaamis- tai ulostusongelmia, jalkojen turvotusta (pojilla kivespussi turpoaa).
Lapsilla mediastinaalinen neuroblastooma (mediastinaalinen neuroblastooma) painaa usein yläonttolaskimoa, mikä voi aiheuttaa kasvojen, kaulan, käsivarsien ja rintakehän turvotusta (iho muuttuu sinertävän punaiseksi ja ihonalaisia kyhmyjä). On yskää ja hengityksen vinkumista, hengitysvaikeuksia (hengenahdistuksen muodossa) tai nielemisvaikeuksia (dysfagia); imusolmukkeet ovat lisääntyneet kaulassa, solisluun yläpuolella, kainaloissa.
Kasvainsolujen leviäminen luuytimeen johtaa anemiaan, trombosytopeniaan ja leukopeniaan, jolla on taipumusta vuotaa verta.
Ja periorbitaalialueen metastaasien yhteydessä silmien ympärille ilmestyy tummia ympyröitä tai mustelmia. Tällainen kasvain voi myös aiheuttaa päänsärkyä ja huimausta, eksoftalmiaa (silmämunien ulkoneminen) ja hermopäätteiden puristumisesta johtuen silmäluomien roikkumista (ptoosi) ja pupillien koon pienenemistä (mioosi).
Vatsan neuroblastooma tai vatsan neuroblastooma lapsilla johtaa tunnustettavien sinettien muodostumiseen vatsaan, turvotukseen, ruokahaluttomuuteen, ummetukseen ja verenpaineen nousuun. Selkäydintä tai hermojuurta painava kasvain voi johtaa raajojen tunnottomuuteen ja heikkouteen, kyvyttömyyteen seistä, ryömiä tai kävellä. Jos luut kärsivät, luukipua voi esiintyä.
Vaiheen 3-4 kasvaimella vatsaontelossa, jossa on vaurioita imusolmukkeissa, kasvainsolut voivat päästä munuaisen parenkyymiin, ja sitten lapsille kehittyy laaja munuaisen neuroblastooma, mikä johtaa sen toimintojen rikkomiseen.
Vaiheet
- Vaiheen 1 neuroblastooma on primaarinen kasvain, joka on lokalisoitu ja eristetty yhdelle kehon alueelle; kummankaan puolen imusolmukkeet eivät vaikuta.
- Neuroblastooma vaihe 2. Vaiheessa 2A primaarinen kasvain rajoittuu yhdelle alueelle, mutta on suuri; kahdenväliset imusolmukkeet eivät vaikuttaneet. Vaiheessa 2B imusolmukkeet sillä kehon puolella, jossa kasvain sijaitsee, ovat positiivisia etäpesäkkeiden suhteen.
- Neuroblastooman vaihe 3: primaarinen kasvain ylittää selkäytimen alueen tai kehon keskiviivan, imusolmukkeista löytyy yksi- tai molemminpuolisia etäpesäkkeitä.
- Vaiheen 4 neuroblastooma: Kasvain on levinnyt kaukaisiin imusolmukkeisiin, luuytimeen, luihin, maksaan tai muihin elimiin. Ja vaihe 4S määritetään alle vuoden ikäisillä lapsilla, joilla on paikallinen primaarinen kasvain, joka leviää ihoon, maksaan tai luuytimeen.
Kansainvälinen neuroblastoomariskin vaiheistusjärjestelmä (INRGSS)
INRGSS käyttää kuvantamisen määrittelemiä riskitekijöitä (IDRF), jotka ovat kuvantamistesteissä havaittuja tekijöitä, jotka voivat tarkoittaa, että kasvain on vaikeampi poistaa.
INRGSS jakaa neuroblastoomat 4 vaiheeseen:
- L1: Kasvain ei ole levinnyt lähtöpaikastaan eikä kasvanut elintärkeiksi rakenteiksi. Se rajoittuu yhteen kehon osaan, kuten kaulaan, rintakehään tai vatsaan.
- L2: Kasvain ei ole levinnyt (metastasoitunut) kauas lähtöpaikastaan (se on esimerkiksi saattanut kasvaa vatsan vasemmalta puolelta rintakehän vasemmalle puolelle), mutta siinä on vähintään yksi IDRF.
- M: Kasvain on metastasoitunut kaukaiseen kehon osaan (paitsi MS-vaiheen kasvaimia).
- MS: Alle 18 kuukauden ikäisten lasten metastaattinen sairaus, jossa syöpä on levinnyt vain ihoon, maksaan ja/tai luuytimeen.
Komplikaatiot ja seuraukset
Neuroblastoomalle on ominaista sellaiset komplikaatiot ja seuraukset kuin:
- leviäminen (metastaasi) imusolmukkeisiin, luuytimeen, maksaan, ihoon ja luihin;
- selkäytimen puristus (joka voi aiheuttaa kipua ja johtaa halvaukseen);
- paraneoplastisen oireyhtymän kehittyminen (johtuen tiettyjen kasvaimen erittämien kemikaalien vaikutuksesta sekä sen solujen ilmentämästä GD2-disialogangliosidiantigeenistä), jotka ilmenevät nopeina tahattomina silmien liikkeinä, koordinaatiohäiriönä, lihaskrampina, ripulina;
- uusiutuminen primaarisen hoidon päätyttyä (kuten kliininen käytäntö osoittaa, suuren riskin neuroblastoomat uusiutuvat 50 %:ssa tapauksista).
Diagnostiikka neuroblastooma
Epäillyn neuroblastooman diagnoosi lapsella vaatii fyysistä tutkimusta, laboratoriotutkimuksia ja kuvantamista.
Veri- ja virtsakokeet otetaan katekoliamiinien (norepinefriini ja dopamiini) ja homovaani- tai vanillyylimantelihapojen (jota muodostuu näiden hormonien aineenvaihdunnan aikana); verikoe neurospesifiselle enolaasille, entsyymi-immunosorbenttimääritys (ELISA) veriseerumista ja luuydintesti (josta otetaan näyte aspiraatiopunktiolla). Mutaatioiden määrittämiseksi tehdään DNA-testi ja kasvainkudoksen sytomorfologista tutkimusta varten tehdään biopsia.
Kun biopsianäytteet on otettu, ne lähetetään laboratorioon, jossa patologi (syöpäsolujen havaitsemiseen erityisesti koulutettu lääkäri) tutkii ne mikroskoopilla. Näytteille tehdään usein myös erityisiä laboratoriotestejä sen osoittamiseksi, onko kasvain neuroblastooma.
Jos kyseessä on neuroblastooma, laboratoriotestit voivat myös auttaa määrittämään, kuinka nopeasti kasvain voi kasvaa tai levitä, sekä mitkä hoidot voivat toimia parhaiten.
Instrumentaalinen diagnostiikka visualisoi kasvaimen ultraäänellä, röntgenillä, MRI:llä tai CT:llä, PET:llä 18F-fluorodeoksiglukoosilla tai MIBG-skannaus - scintigrafia metaiodobentsyyliguanidiinilla. [1]
Differentiaalinen diagnoosi
Erotusdiagnoosi sisältää hyvänlaatuisen ganglioneurooman, ganglioneuroblastooman. Rabdomyosarkooma, nefroblastooma.
Kuka ottaa yhteyttä?
Hoito neuroblastooma
Neuroblastoomassa hoito riippuu potilaan riskiryhmästä (kasvainprosessin vaiheesta), kasvaimen sijainnista, kasvainsolujen genomista ominaisuuksista ja lapsen iästä. Ja se voi sisältää seurantaa, leikkausta, kemoterapiaa, sädehoitoa, immunoterapiaa, hematopoieettisten kantasolujen siirtoa .
Neoadjuvantti tai adjuvantti (pre- tai postoperatiivinen) kemoterapia lasten neuroblastoomaan, kuten mikä tahansa syövän kemoterapia , suoritetaan kursseilla: lääkettä annetaan useita päiviä peräkkäin, minkä jälkeen pidetään tauko kehon palauttamiseksi. Syklit toistuvat yleensä kolmen tai neljän viikon välein.
Käytetään seuraavia lääkkeitä (ja niiden yhdistelmää): syklofosfamidi, sisplatiini tai karboplatiini, doksorubisiini (adriamysiini), vinkristiini, etoposidi.
Kemoterapialääkkeiden yleisiä sivuvaikutuksia ovat hiustenlähtö, ruokahaluttomuus, lisääntynyt väsymys, pahoinvointi ja oksentelu, suun haavaumat, ripuli tai ummetus. Kemoterapia voi vaikuttaa negatiivisesti luuytimeen ja aiheuttaa verisolujen määrän vähenemisen.
Kohdennettussa immunoterapiassa (joka on suunnattu GD2-kasvainantigeeniin) käytetään monoklonaalisten vasta-aineiden (anti-GD2 MAb) lääkkeitä Dinutuximab (Unituxin) ja Naxitamab. Ne annetaan suonensisäisesti jatkuvana infuusiona yhdessä granulosyytti-makrofagipesäkkeitä stimuloivan tekijän (GM-CSF-sytokiinin) ja interleukiini-2:n kanssa.
Näiden lääkkeiden sivuvaikutukset ilmenevät kivuna (usein erittäin vaikeana), verenpaineen laskuna, sydämentykytysnä, hengenahdistuksena (mahdollisesti hengitysteiden turvotuksena), kuumeina, pahoinvointina, oksenteluna ja ripulina, solu- ja mineraalimuutoksina. Veren koostumus.
Syövän uusiutumisen riskin vähentämiseksi suuriannoksisen kemoterapian ja kantasolusiirron jälkeen lapsia, joilla on korkean riskin neuroblastooma, hoidetaan systeemisillä retinodeilla, 13-cis-retinoiinihapolla (isotretinoiinilla). [2]
Neuroblastooman kirurginen hoito - kasvaimen poistaminen, esimerkiksi avoin lisämunuaisen poisto tai lisämunuaisen neuroblastooman laparoskooppinen resektio; lymfedektomia (vaurioituneiden imusolmukkeiden poistaminen) jne. [3]
Korkean riskin neuroblastooman tapauksessa voidaan antaa sädehoitoa . [4]
Ennaltaehkäisy
Kun otetaan huomioon lasten neuroblastooman syyt, ainoa tapa estää se voi olla geneettinen neuvonta raskautta suunniteltaessa. Mutta on pidettävä mielessä, että tämä kasvain liittyy perinnöllisiin mutaatioihin vain 1-2 prosentissa tapauksista.
Ennuste
Vauvan neuroblastoomalla on kyky taantua spontaanisti.
Prognostiset merkit
- Korkean riskin kasvaimilla sekä neuroblastoomalla kaikissa ikäryhmissä ja kaikissa vaiheissa (paitsi vaihe 4S) - lisääntyneellä N-MYC-geenin ilmentymisellä ja N-Myc-onkogeenin lisääntymisellä - on epäsuotuisa ennuste, joka vaikuttaa elinajanodotteeseen.
- Kasvainsolujen esiintyminen, joista puuttuu tiettyjä osia kromosomista 1 tai 11 (tunnetaan nimellä 1p- tai 11q-deleetiot), antaa vähemmän suotuisan ennusteen. Kromosomin 17 ylimääräinen osa (lisäys 17q:ssa) liittyy myös huonompaan ennusteeseen.
- Neuroblastoomasoluilla, joissa on enemmän DNA:ta, on parempi ennuste, erityisesti alle 2-vuotiailla lapsilla.
- Neuroblastoomilla, joissa on enemmän neurotrofiinireseptoreita, erityisesti hermokasvutekijäreseptoria TrkA, on suotuisampi ennuste.
Eloonjääminen riskiryhmään kuuluvan lapsuuden syöpäryhmän (COG) mukaan
- Pienen riskin ryhmä: Matalan riskin ryhmän lasten 5 vuoden eloonjäämisaste on yli 95%.
- Keskitason riskiryhmä: Keskitason riskiryhmän lasten 5 vuoden eloonjäämisaste on 90–95 %.
- Korkean riskin ryhmä: Korkean riskin lasten 5 vuoden eloonjäämisaste on noin 50%.
Noin 15 % lasten syöpäkuolemista liittyy neuroblastoomaan. Tällä suuren riskin pahanlaatuisella kasvaimella pitkän aikavälin eloonjäämismahdollisuudet eivät ylitä 40%. Viiden vuoden kokonaiseloonjäämisaste on 67-74 %, ikäryhmässä 1-4 vuotta - 43 % ja ensimmäisen elinvuoden aikana diagnosoidun neuroblastooman osalta yli 80 %.
Использованная литература