Lääketieteen asiantuntija

Uudet julkaisut
Neuroblastooma lapsilla: syyt, diagnoosi, hoito
Viimeksi tarkistettu: 04.07.2025

Kaikki iLive-sisältö tarkistetaan lääketieteellisesti tai se tarkistetaan tosiasiallisen tarkkuuden varmistamiseksi.
Meillä on tiukat hankintaohjeet ja vain linkki hyvämaineisiin mediasivustoihin, akateemisiin tutkimuslaitoksiin ja mahdollisuuksien mukaan lääketieteellisesti vertaisarvioituihin tutkimuksiin. Huomaa, että suluissa ([1], [2] jne.) Olevat numerot ovat napsautettavia linkkejä näihin tutkimuksiin.
Jos sinusta tuntuu, että jokin sisältö on virheellinen, vanhentunut tai muuten kyseenalainen, valitse se ja paina Ctrl + Enter.
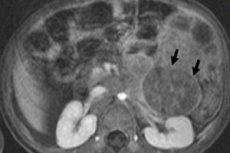
Lasten onkologiassa yksi yleisimmistä kallon ulkopuolisista kasvaimista on neuroblastooma, joka on hermosoluhuippujen neuroblastien eli sympaattisen hermoston alkion (epäkypsien) hermosolujen alkion pahanlaatuinen kasvain.
Epidemiologia
Kansainvälisen neuroblastoomariskiryhmän (INRG) tilastojen mukaan neuroblastooma on maailmanlaajuinen syy noin 8 prosenttiin kaikista lasten onkologisista sairauksista ja on esiintyvyydessä kolmanneksi yleisin leukemian ja aivokasvainten jälkeen.
Muiden tietojen mukaan neuroblastooma muodostaa noin 28 % kaikista imeväisten syöpätyypeistä. Yli kolmannes neuroblastoomatapauksista diagnosoidaan alle vuoden ikäisillä lapsilla; diagnoosin keskimääräinen ikä on 19–22 kuukautta. Yli 90 % diagnosoiduista tapauksista esiintyy 2–5-vuotiailla lapsilla (poikien ollessa vallitsevia); ilmaantuvuuden huippu havaitaan 2–3 vuoden iässä, ja yli viisivuotiailla lapsilla tapausten osuus on alle 10 %.
Syyt neuroblastoomat
Tutkiessaan neuroblastooman syitä tutkijat ovat tulleet siihen tulokseen, että tämä kasvain lapsilla esiintyy satunnaisten geneettisten mutaatioiden seurauksena alkionkehityksen tai varhaisen postnataalisen kehityksen aikana. Mutta näiden geenimuutosten syy on tuntematon, koska teratogeenisten ympäristötekijöiden vaikutusta ei ole tunnistettu.
Näitä kasvaimia voi esiintyä missä tahansa, mukaan lukien välikarsina, kaula, vatsa, lisämunuaiset, munuaiset, selkäranka ja lantio.
Harvinaisissa tapauksissa imeväisten neuroblastoomaan voi liittyä perinnöllinen mutaatio. Erityisesti mutaatio kalvoproteiini CD246:n geenissä kromosomissa 2 – tyrosiinikinaasientsyymissä ALK, joka varmistaa solujen välisen viestinnän ja jolla on tärkeä rooli hermoston toiminnassa; tai PHOX2B-proteiinin geenissä (kromosomissa 4), joka osallistuu hermosolujen kypsymiseen.
Neuroblastooma voi liittyä myös lapsuusiän neurofibromatoosiin tyyppiä 1,Beckwith-Wiedemannin oireyhtymään ja hyperinsulinemiseen hypoglykemiaan (nesidioblastoosipankreatiitti).
Riskitekijät
Nykyään perinnöllisyys tunnustetaan lasten neuroblastooman kehittymisen riskitekijöiksi - kasvaimen esiintyminen suvussa sekä synnynnäiset poikkeavuudet, jotka liittyvät geenimutaatioihin kohdunsisäisen kehityksen aikana. Tämä pätee erityisesti tapauksiin, joissa useiden kasvainten kehittymistä eri elimissä.
Tutkijat eivät ole tunnistaneet mitään tämän kasvaimen riskiä lisäävistä ulkoisista tekijöistä.
Synnyssä
Neuroblastoomien kehitysmekanismi johtuu hermosoluharjasolujen – ihmisalkion ektodermaalisesta alkiokerroksesta hermoputken reunoille muodostuvien kahdenvälisten solulinjojen – erilaistumisen ja kypsymisen häiriöistä. Nämä solut vaeltavat (liikkuvat) ja erilaistuvat moniksi solutyypeiksi: sensorisiksi ja autonomisiksi neuroneiksi, neuroendokriinisiksi soluiksi ja lisämunuaisen ytimen soluiksi, kallon ja kasvojen ruston ja luiden soluiksi sekä pigmenttisoluiksi.
Neuroblastoomassa siirtyneet neuroblastit eivät kypsy, vaan jatkavat kasvuaan ja jakautumista muodostaen kasvaimen. Sen muodostumisen patogeneesi liittyy seuraaviin geenimutaatioihin:
- kromosomisekvenssin osan päällekkäisyydellä tai LMO1-geenin segmenttien päällekkäisyydellä kromosomissa 11, joka koodaa RBTN1-proteiinia alkion hermosoluissa;
- ihmisen hermostollisten kantasolujen lisääntymistä säätelevän DUF1220-proteiinia koodaavan NBPF10-geenin kopiomäärän muutos kromosomissa 1q21.1. Nämä häiriöt johtavat joko tämän kromosomin kahdentumiseen tai sen deleetioon – osan DNA:sta puuttumiseen;
- muutoksilla kasvainsuppressorigeenissä ATRX (kromosomissa Xq21.1);
- N-Myc-transkriptiotekijägeenin lisäkopioiden (amplifikaation) läsnäololla kromosomissa 2, joka koodaa yhtä transkriptiotekijöistä (DNA:ta sitova proteiini), joka säätelee muiden geenien aktiivisuutta ja kontrolloi esiastesolujen lisääntymistä sikiön kudosten ja elinten muodostumiseen tarvittavien proteiinien muodostumisen aikana. Tämän geenin amplifikaatio muuttaa sen onkogeeniksi, mikä aiheuttaa solusyklin häiriintymisen, lisääntyneen solujen lisääntymisen ja kasvainten muodostumisen.
Oireet neuroblastoomat
Neuroblastooman ensimmäiset oireet ovat epäspesifisiä ja niihin voivat kuulua ruokahaluttomuus (ja painonpudotus), väsymys syötettäessä, kuume ja nivelkipu.
Kliiniset oireet riippuvat primaarituumorin sijainnista ja etäpesäkkeiden esiintymisestä (joita esiintyy 60–73 %:ssa tapauksista).
Hyvin usein primaarinen neuroblastooma lokalisoituu lisämunuaisen ytimessä, jolla on samanlainen alkuperä kuin hermosoluilla. Alle vuoden ikäisillä lapsilla lisämunuaisen neuroblastoomaa diagnosoidaan 35–40 %:ssa tapauksista. Sen oireita ovat vatsakipu, kuume, painon lasku, luukipu, anemia tai samanaikainen Pepperin oireyhtymä: diffuusi maksavaurio, johon liittyy vaikea hepatomegalia ja hengitysvaikeusoireyhtymä.
Retroperitoneaalinen neuroblastooma eli retroperitoneaalinen neuroblastooma lapsilla alkaa kasvaessaan painaa virtsarakkoa tai suolistoa, mikä voi aiheuttaa virtsaamis- tai ulostamisongelmia, jalkojen turvotusta (pojilla kivespussi turpoaa).
Lapsilla esiintyvä mediastinum-neuroblastooma (mediastinaalinen neuroblastooma) painaa usein yläonttolaskimoa, ja tämä voi aiheuttaa kasvojen, kaulan, käsivarsien ja ylärintakehän turvotusta (iho muuttuu sinertävän punaiseksi ja ihonalaisiin kyhmyihin). Esiintyy yskää ja hengityksen vinkumista, hengitysvaikeuksia (hengenahdistusta) tai nielemisvaikeuksia (dysfagiaa); suurentuneita imusolmukkeita havaitaan kaulassa, solisluun yläpuolella ja kainaloissa.
Kasvainsolujen leviäminen luuytimeen johtaa anemiaan, trombosytopeniaan ja leukopeniaan, joilla on taipumus verenvuotoon.
Ja silmäkuopan ympärillä olevien etäpesäkkeiden yhteydessä silmien ympärille ilmestyy tummia silmänalusia tai mustelmia. Tällainen kasvain voi aiheuttaa myös päänsärkyä ja huimausta, silmämunien pullistumaa (exophthalmiaa) ja hermopäätteiden puristumisesta johtuen roikkuvia silmäluomia (ptoosia) ja pupillien pienenemistä (mioosia).
Vatsan neuroblastooma eli vatsaontelon neuroblastooma lapsilla johtaa tunnusteltavien tiivisteiden muodostumiseen vatsaan, sen laajenemiseen, ruokahaluttomuuteen, ummetukseen ja verenpaineen nousuun. Selkäydintä tai hermojuurta painava kasvain voi johtaa raajojen tunnottomuuteen ja heikkouteen, kyvyttömyyteen seistä, ryömiä tai kävellä. Jos luut vaurioituvat, voi esiintyä luukipua.
Jos vatsaontelossa on vaiheen 3-4 kasvain, johon liittyy imusolmukkeiden vaurioita, kasvainsolut voivat päästä munuaisten parenkyymiin, ja sitten lapsilla kehittyy laaja munuaisen neuroblastooma, joka johtaa sen toiminnan häiriintymiseen.
Vaiheet
- Vaiheen 1 neuroblastooma on primaarituumori, joka on paikallinen ja eristyksissä kehon yhteen alueeseen; molempien puolien imusolmukkeet eivät ole vaurioituneet.
- Neuroblastooma, vaihe 2. Vaiheessa 2A primaarituumori rajoittuu yhteen alueeseen, mutta on suuri; molemminpuoliset imusolmukkeet eivät ole levinneet. Vaiheessa 2B kasvaimen sijaintipuolen imusolmukkeet ovat etäpesäkkeitä.
- Neuroblastooman vaihe 3: primaarituumori ylittää selkäytimen tai kehon keskiviivan, imusolmukkeissa on yksi- tai kahdenvälisiä etäpesäkkeitä.
- Neuroblastooman vaihe 4: kasvain on levinnyt kaukaisiin imusolmukkeisiin, luuytimeen, luihin, maksaan tai muihin elimiin. Vaihe 4S määritetään alle vuoden ikäisillä lapsilla, joilla on paikallinen primaarituumori, joka on levinnyt ihoon, maksaan tai luuytimeen.
Kansainvälinen neuroblastoomariskien luokitusjärjestelmä (INRGSS)
INRGSS käyttää kuvantamisen avulla määriteltyjä riskitekijöitä (IDRF), jotka ovat kuvantamistutkimuksissa havaittavia tekijöitä, jotka voivat vaikeuttaa kasvaimen poistamista.
INRGSS jakaa neuroblastoomat neljään vaiheeseen:
- L1: Kasvain ei ole levinnyt lähtökohdastaan eikä kasvanut elintärkeiksi rakenteiksi. Se rajoittuu yhteen kehon osaan, kuten kaulaan, rintaan tai vatsaan.
- L2: Kasvain ei ole levinnyt (metastasoitunut) kauas lähtökohdastaan (se on esimerkiksi saattanut kasvaa vatsan vasemmalta puolelta rintakehän vasemmalle puolelle), mutta sillä on vähintään yksi IDRF.
- M: Kasvain on etäpesäkkeistynyt kaukaiseen kehon osaan (lukuun ottamatta MS-vaiheen kasvaimia).
- MS: Alle 18 kuukauden ikäisillä lapsilla esiintyvä etäpesäkkeinen sairaus, jossa syöpä on levinnyt vain ihoon, maksaan ja/tai luuytimeen.
Komplikaatiot ja seuraukset
Neuroblastoomalle on ominaista komplikaatiot ja seuraukset, kuten:
- leviäminen (metastaasi) imusolmukkeisiin, luuytimeen, maksaan, ihoon ja luihin;
- selkäytimen puristus (joka voi aiheuttaa kipua ja johtaa halvaantumiseen);
- paraneoplastisen oireyhtymän kehittyminen (johtuen tiettyjen kasvaimen erittämien kemikaalien vaikutuksesta sekä sen solujen ilmentämästä antigeenistä disialogangliosidi GD2), joka ilmenee nopeina tahattomina silmänliikkeinä, heikentyneenä koordinaationa, lihaskramppeina ja ripulina;
- relapsit primaarihoidon päättymisen jälkeen (kuten kliininen käytäntö osoittaa, suuririskisillä neuroblastoomilla on relapsi 50 %:ssa tapauksista).
Diagnostiikka neuroblastoomat
Epäillyn neuroblastooman diagnosointi lapsella edellyttää tutkimusta, laboratoriokokeita ja kuvantamista.
Veri- ja virtsakokeista tutkitaan katekoliamiineja (noradrenaliini ja dopamiini) sekä homovanilliini- tai vanillyylimantelihappoja (joita muodostuu näiden hormonien aineenvaihdunnassa); verikokeesta määritetään neurospesifinen enolaasi, veriseerumista tehdään entsyymi-immunosorbenttimääritys (ELISA) ja luuydinnäyte otetaan imupunktiolla. Mutaatioiden määrittämiseksi tehdään DNA-testi ja kasvainkudoksen sytomorfologista tutkimusta varten otetaan koepala.
Kun koepalanäytteet on otettu, ne lähetetään laboratorioon, jossa patologi (lääkäri, jolla on erityiskoulutus syöpäsolujen tunnistamiseen) tutkii niitä mikroskoopilla. Näytteille tehdään usein myös erityisiä laboratoriotestejä sen selvittämiseksi, onko kasvain neuroblastooma.
Jos kyseessä on neuroblastooma, laboratoriotestit voivat myös auttaa määrittämään, kuinka nopeasti kasvain voi kasvaa tai levitä, sekä mitkä hoidot saattavat toimia parhaiten.
Instrumentaalinen diagnostiikka visualisoi kasvaimen ultraäänellä, röntgenillä, magneettikuvauksella tai tietokonetomografialla, PET-kuvauksella ja 18F-fluorodeoksiglukoosi-infuusiolla tai MIBG-skannauksella - skintigrafialla ja metajodobentsyyliguanidiinilla. [ 1 ]
Differentiaalinen diagnoosi
Differentiaalidiagnoosiin kuuluvat hyvänlaatuinen ganglioneuroma, ganglioneuroblastooma, rabdomyosarkooma ja nefroblastooma.
Kuka ottaa yhteyttä?
Hoito neuroblastoomat
Neuroblastoomassa hoito riippuu potilaan riskiryhmästä (kasvainprosessin vaiheesta), kasvaimen sijainnista, kasvainsolujen genomisista ominaisuuksista ja lapsen iästä. Se voi sisältää seurantaa, leikkausta, kemoterapiaa, sädehoitoa, immunoterapiaa ja hematopoieettisten kantasolujen siirtoa.
Neoadjuvantti eli adjuvantti (pre- tai postoperatiivinen) kemoterapia lasten neuroblastoomaan, kuten mikä tahansa syöpäkemoterapia, annetaan kuureina: lääkettä annetaan useita päiviä peräkkäin, minkä jälkeen pidetään tauko, jotta keho voi toipua. Syklit toistetaan yleensä 3–4 viikon välein.
Seuraavia lääkkeitä (ja niiden yhdistelmiä) käytetään: syklofosfamidi, sisplatiini tai karboplatiini, doksorubisiini (adriamysiini), vinkristiini, etoposidi.
Kemoterapialääkkeiden yleisiä sivuvaikutuksia ovat hiustenlähtö, ruokahaluttomuus, väsymys, pahoinvointi ja oksentelu, suun haavaumat, ripuli tai ummetus. Kemoterapia voi vahingoittaa luuydintä ja aiheuttaa verisolujen määrän vähenemistä.
Kohdennettu immunoterapia (kohdistettu kasvainantigeeniin GD2) käyttää monoklonaalisten vasta-aineiden (anti-GD2 MAb) ryhmään kuuluvia lääkkeitä, dinutuksimabia (Unituxin) ja naksitamabia. Ne annetaan laskimonsisäisenä pitkäkestoisena infuusiona yhdessä granulosyytti-makrofagikasvutekijän (sytokiini GM-CSF) ja interleukiini-2:n kanssa.
Näiden lääkkeiden sivuvaikutuksia ovat kipu (usein erittäin voimakas), verenpaineen lasku, sydämen sykkeen nousu, hengenahdistus (mahdollisesti hengitysteiden turvotuksen kanssa), kuume, pahoinvointi, oksentelu ja ripuli sekä muutokset veren solu- ja mineraalikoostumuksessa.
Suuriannoksisen kemoterapian ja kantasolusiirron jälkeisen syövän uusiutumisriskin vähentämiseksi lapsilla, joilla on suuririskinen neuroblastooma, annetaan systeemisiä retinoideja, 13-cis-retinoiinihappoa (isotretinoiinia). [ 2 ]
Neuroblastooman kirurginen hoito – kasvaimen poisto, esimerkiksi avoin adrenalektomia tai laparoskooppinen lisämunuaisen neuroblastooman resektio; lymfektomia (sairauden kohteeksi joutuneiden imusolmukkeiden poisto) jne. [ 3 ]
Korkean riskin neuroblastoomassa voidaan käyttää sädehoitoa.[ 4 ]
Ennaltaehkäisy
Ottaen huomioon neuroblastooman syyt lapsilla, ainoa ennaltaehkäisevä toimenpide voi olla geneettinen neuvonta raskautta suunniteltaessa. On kuitenkin pidettävä mielessä, että tämä kasvain liittyy perinnöllisiin mutaatioihin vain 1-2 prosentissa tapauksista.
Ennuste
Infantiilisella neuroblastoomalla on kyky taantua itsestään.
Ennusteelliset markkerit
- Korkean riskin kasvaimet, samoin kuin kaikenikäisten ja -vaiheisten lasten neuroblastoomat (paitsi vaihe 4S) – joihin liittyy N-MYC-geenin ilmentymisen lisääntyminen ja N-Myc-onkogeenin monistuminen – ennustavat huonosti ja vaikuttavat elinajanodotteeseen.
- Kasvainsoluilla, joista puuttuu tiettyjä osia kromosomeista 1 tai 11 (tunnetaan 1p- tai 11q-deleetioina), on huonompi ennuste. Myös ylimääräinen osa kromosomista 17 (17q:n lisäys) liittyy huonompaan ennusteeseen.
- Neuroblastoomasoluilla, joilla on suuri määrä DNA:ta, on parempi ennuste, erityisesti alle 2-vuotiailla lapsilla.
- Neuroblastoomilla, joilla on enemmän neurotrofiinireseptoreita, erityisesti hermokasvutekijäreseptori TrkA, on parempi ennuste.
Eloonjääminen lapsuuden onkologian ryhmän (COG) riskiryhmän mukaan
- Alhaisen riskin ryhmä: Alhaisen riskin ryhmän lapsilla on 5 vuoden eloonjäämisaste yli 95 %.
- Keskivaikea riskiryhmä: Keskivaikeaan riskiryhmään kuuluvien lasten 5 vuoden eloonjäämisaste on 90–95 %.
- Korkean riskin ryhmä: Korkean riskin ryhmään kuuluvien lasten 5 vuoden eloonjäämisaste on noin 50 %.
Noin 15 % lapsuusiän syöpäkuolemista johtuu neuroblastoomasta. Tämän riskialttiimman pahanlaatuisen kasvaimen pitkäaikaiselossaolomahdollisuudet ovat enintään 40 %. Viiden vuoden kokonaiselossaoloaste on 67–74 %, 43 % 1–4-vuotiailla ja yli 80 % ensimmäisenä elinvuotena diagnosoidulla neuroblastoomalla.
Использованная литература