
Charcot-Marie-Tooth-tauti
Viimeksi tarkistettu: 23.11.2021

Kaikki iLive-sisältö tarkistetaan lääketieteellisesti tai se tarkistetaan tosiasiallisen tarkkuuden varmistamiseksi.
Meillä on tiukat hankintaohjeet ja vain linkki hyvämaineisiin mediasivustoihin, akateemisiin tutkimuslaitoksiin ja mahdollisuuksien mukaan lääketieteellisesti vertaisarvioituihin tutkimuksiin. Huomaa, että suluissa ([1], [2] jne.) Olevat numerot ovat napsautettavia linkkejä näihin tutkimuksiin.
Jos sinusta tuntuu, että jokin sisältö on virheellinen, vanhentunut tai muuten kyseenalainen, valitse se ja paina Ctrl + Enter.
Peroneaalisen lihaksen atrofia, oireyhtymä tai Charcot-Marie-Tooth-tauti on koko ryhmä kroonisia perinnöllisiä sairauksia, joilla on perifeeristen hermojen vaurioita.
ICD-10: n mukaan hermoston sairauksien osassa tämän taudin koodi on G60.0 (perinnöllinen motorinen ja sensorinen neuropatia). Se sisältyy myös harvinaislääkkeiden luetteloon.
Epidemiologia
Kliinisten tilastojen mukaan kaiken tyyppisten Charcot-Marie-Tooth-tautien esiintyvyys 100 tuhatta väestöä kohti on 19 tapausta (muiden lähteiden mukaan yksi tapaus 2,5-10 tuhatta väestöä kohti).
CMT-tyypin 1 osuus on noin kaksi kolmasosaa tapauksista (yksi tapaus 5-7 tuhatta väestöä kohden), ja melkein 70% niistä liittyy PMP22-geenin kaksoiskappaleisiin. Maailmassa tällainen sairaus vaikuttaa yli 1,2 miljoonaan ihmiseen.
Tyypin 4 CMT: n ilmaantuvuuden arvioidaan olevan 1-5 tapausta 10 tuhatta lasta kohti. [1]
Syyt charcot-Marie-Tooth-tauti
Mukaan luokittelu polyneuropathic oireyhtymien , peroneal (peroneal) lihasten surkastumista, Charcot-Marie-Tooth neuraalisen amyotrofiakompleksia tai Charcot-Marie-Toothin tauti (lyhennetään CMT) tarkoitetaan geneettisesti määräytyvä moottori aisteihin polyneuropatioiden. [2]
Toisin sanoen syyt sen esiintymiseen ovat geneettiset mutaatiot. Ja geneettisten poikkeavuuksien luonteesta riippuen tämän oireyhtymän päätyypit tai tyypit eroavat toisistaan: demyelinoiva ja aksonaalinen. Ensimmäiseen ryhmään kuuluu tyypin 1 Charcot-Marie-Tooth-tauti (CMT1), joka esiintyy PMP22-geenin monistumisen seurauksena kromosomissa 17, joka koodaa transmembraanista perifeeristä myeliiniproteiinia 22. Tämän seurauksena aksonivaipan segmentaalinen demyelinaatio (hermosolujen prosessit) ja hermoston johtumisnopeus vähenee. Signaalit. Lisäksi joissakin muissa geeneissä voi olla mutaatioita.
Aksonaalimuoto on Charcot-Marie-Toothin taudin tyyppi 2 (CMT2), joka vaikuttaa itse aksoneihin ja liittyy patologisiin muutoksiin MFN2-geenissä 1p36.22-lokuksessa, joka koodaa kalvoproteiinia mitofusiini-2, mikä on välttämätöntä. Mitokondrioiden fuusioon ja toiminnallisten mitokondrioiden muodostumiseen solujen ääreishermoissa. CMT2-alatyyppejä on yli tusina (mutaatioilla spesifisissä geeneissä).
On huomattava, että nyt on tunnistettu yli sata geeniä, joiden vahingot, perittyinä, aiheuttavat erilaisia Charcot-Marie-Tooth-taudin alatyyppejä. Esimerkiksi mutaatiot RAB7-geenissä kehittävät tyypin 2B CMT; SH3TC2-geenin muutos (joka koodaa yhtä Schwannin solukalvojen proteiineista) aiheuttaa tyypin 4C CMT, joka ilmenee lapsuudessa ja jolle on tunnusomaista motoristen ja aistien hermosolujen (puolitoista tusinaa 4 tämä tauti erotetaan).
Harvinainen tyypin 3 SMT (kutsutaan Dejerine-Sott-oireyhtymäksi), jonka aiheuttavat mutaatiot PMP22-, MPZ-, EGR2- ja muissa geeneissä, alkaa kehittyä myös varhaislapsuudessa.
Kun CMT-tyyppi 5 esiintyy 5-12-vuotiaana, ei vain motorista neuropatiaa (alaraajojen spastisen parapareesin muodossa) havaita, vaan myös näkö- ja kuulohermojen vaurioita.
Lihasten heikkous ja näköhermon atrofia (näön menetys) sekä tasapaino-ongelmat ovat tyypillisiä CMT-tyypille 6. Ja tyypin 7 Charcot-Marie-Tooth-taudin yhteydessä ei havaita vain motorisen aistinvaraisen neuropatian lisäksi verkkokalvon tautia retinitis pigmentosan muodossa.
Miesten yleisempi X-sidottu SMT- tai Charcot-Marie-Tooth-tauti, johon liittyy raajojen tetrapareesi (molempien käsivarsien ja jalkojen liikkumisen heikkeneminen), on demyelinoiva tyyppi, ja sitä pidetään GJB1-geenin mutaation seurauksena X-kromosomin pitkä käsivarsi, joka koodaa konnexiinia 32, transmembraanista proteiinia Schwann-soluja ja oligodendrosyyttejä, joka säätelee hermosignaalien siirtymistä. [3]
Riskitekijät
CMT: n tärkein riskitekijä on tämän taudin esiintyminen suvussa, toisin sanoen lähisukulaisissa.
Geneetikkojen mukaan, jos molemmat vanhemmat ovat Charcot-Marie-Toothin taudin autosomaalisen resessiivisen geenin kantajia, riski saada lapsi, jolle kehittyy tämä tauti, on 25%. Ja riskin, että lapsi kantaa tätä geeniä (mutta hänellä itsellään ei ole oireita), arvioidaan olevan 50%.
X-liitännäisen perinnön tapauksessa (kun mutatoitunut geeni on naisen X-kromosomissa), on 50% riski, että äiti siirtää tämän geenin pojalleen, ja hänelle kehittyy CMT-tauti. Kun naislapsi syntyy, tautia ei ehkä esiinny, mutta tyttären pojat (lapsenlapset) voivat periä viallisen geenin - taudin kehittyessä.
Synnyssä
Minkä tahansa tyyppisessä Charcot-Marie-Tooth-taudissa sen patogeneesi johtuu perifeeristen hermojen perinnöllisestä poikkeavuudesta: moottori (motorinen) ja aistien (aistinvarainen).
Jos CMT-tyyppi on demyelinoiva, niin myeliinivaipan tuhoutuminen tai vika, joka suojaa perifeeristen hermojen aksoneja, johtaa perifeerisen hermoston hermoimpulssien siirtymisen hidastumiseen - aivojen, lihasten ja aistielinten välillä.
Taudin aksonaalisessa tyypissä aksonit vaikuttavat suoraan, mikä vaikuttaa negatiivisesti hermosignaalien voimakkuuteen, mikä ei riitä lihasten ja aistielinten täydelliseen stimulointiin.
Lue myös:
Kuinka Charcot-Marie-Tooth -oireyhtymä leviää? Vialliset geenit voidaan periä autosomaalisesti hallitsevalla tai autosomaalisella resessiivisellä tavalla.
Yleisin - autosomaalinen hallitseva perintö - tapahtuu, kun mutatoidusta geenistä on yksi kopio (yhden vanhemman kantama). CMT: n leviämisen todennäköisyyden jokaiselle syntyneelle lapselle arvioidaan 50 prosentiksi. [4]
Autosomaalisen resessiivisen perinnön kohdalla tauti vaatii kaksi kopiota viallisesta geenistä (yhden kustakin vanhemmasta, jolla ei ole merkkejä taudista).
40-50 prosentissa tapauksista tapahtuu autosomaalisesti hallitseva perinnöllinen demyelinaatio, ts. CMT-tyyppi 1; 12-26% tapauksista - aksonaalinen CMT, eli tyyppi 2. Ja 10-15 prosentissa tapauksista havaitaan X-sidottu perintö. [5]
Oireet charcot-Marie-Tooth-tauti
Yleensä tämän taudin ensimmäiset merkit alkavat näkyä lapsuudessa ja nuorilla ja kehittyvät vähitellen koko elämän ajan, vaikka oireyhtymä voi tuntua myöhemmin. Oireiden yhdistelmä on vaihteleva, eikä taudin etenemisnopeutta eikä sen vakavuutta voida ennustaa.
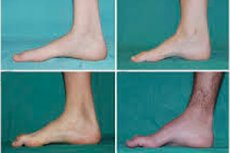
Alkuvaiheessa on sellaisia tyypillisiä oireita kuin lisääntynyt yleinen väsymys; jalkojen, nilkkojen ja säären lihasten heikentynyt sävy (heikkous); refleksien puute. Tämä vaikeuttaa jalan liikuttamista ja johtaa dysbasiaan (kävelyhäiriöihin) jalkojen korkeamman nousun muodossa, usein usein kompastamalla ja kaatumalla. Pienen lapsen Charcot-Marie-Tooth-taudin merkit voivat olla voimakkaita ikään liittyvää kömpelyyttä ja kävelyvaikeuksia, jotka liittyvät kahdenväliseen roikkuvaan jalkaan . Jalkojen epämuodostumat ovat myös ominaisia: korkea kaari (ontto jalka) tai vahvat litteät jalat, kaarevat (vasaramaiset) sormet.
Jos kävelet varpailla lihaksen hypotension taustalla, neurologi voi epäillä, että lapsella on tyypin 4 CMT, jossa lapset eivät murrosiässä kykene kävelemään.
Sen edetessä lihasten surkastuminen ja heikkous leviävät yläraajoihin, mikä vaikeuttaa hienomotoriikkaa ja normaalia käsien toimintaa. Taktiilien väheneminen ja kyky tuntea lämmin ja kylmä sekä jalkojen ja käsien tunnottomuus osoittavat aistihermojen aksonien vaurioitumisen.
Kun se ilmenee tyypin 3 ja 6 lapsuuden Charcot-Marie-Tooth-taudissa, on herkkä ataksia (heikentynyt liikkeen ja tasapainon koordinaatio), lihasten nykiminen ja vapina, kasvohermon vaurio, näköhermon atrofia nystagmilla, kuulonalenema.
Myöhemmissä vaiheissa voi olla hallitsematonta vapinaa (vapinaa) ja usein lihaskramppeja; liikkumisongelmat voivat johtaa kivun kehittymiseen: lihas, nivel, neuropaattinen.
Komplikaatiot ja seuraukset
Charcot-Marie-Tooth -taudilla voi olla komplikaatioita ja seurauksia, kuten:
- useammat nyrjähdykset ja murtumat;
- supistukset, jotka liittyvät periartikulaaristen lihasten ja jänteiden lyhentämiseen;
- skolioosi (selkärangan kaarevuus);
- hengitysvaikeudet - vahingoittamalla hermokuituja, jotka innervoivat kalvon lihaksia:
- menetys kyvystä liikkua itsenäisesti.
Diagnostiikka charcot-Marie-Tooth-tauti
Diagnostiikka sisältää kliinisen tutkimuksen, historian (mukaan lukien sukututkimus), neurologisen ja systeemisen tutkimuksen.
Testit suoritetaan liikealueen, herkkyyden ja jänteen refleksien tarkistamiseksi. Hermoston johtuminen voidaan arvioida instrumentaalidiagnostiikalla - elektromyografialla tai elektroneuromyografialla . Ultraääni tai MRI voidaan myös tarvita. [6]
Geneettinen tai DNA-diagnostiikka yleisimpien geneettisten mutaatioiden tunnistamiseksi, jotka aiheuttavat CMT: tä verinäytteessä, on rajallinen, koska DNA-testejä ei tällä hetkellä ole saatavana kaikentyyppisille CMT: lle. Katso lisätietoja - Geneettinen tutkimus
Joissakin tapauksissa tehdään ääreishermon (yleensä gastrocnemius) biopsia.
Differentiaalinen diagnoosi
Differentiaalinen diagnoosi suoritetaan muiden perifeeristen neuropatioiden, Duchennen lihasdystrofian, myelopaattisten ja myasteenisten oireyhtymien, diabeettisen neuropatian, myeloapioiden kanssa multippelisen ja amyotrofisen lateraaliskleroosin, Guillain-Barré -oireyhtymän, peroneaalisen hermon trauman ja sen kiekkojen atrofian (mukaan lukien selkärangan) yhteydessä ), pikkuaivojen tai talamuksen vauriot sekä kemoterapian sivuvaikutukset (hoidettaessa sytostaateilla, kuten Vincristine tai Paclitaxel). [7]
Kuka ottaa yhteyttä?
Hoito charcot-Marie-Tooth-tauti
Nykyään tämän perinnöllisen taudin hoito koostuu fysioterapiaharjoituksista (joiden tarkoituksena on vahvistaa ja venyttää lihaksia); toimintaterapia (joka auttaa potilaita, joilla on käsien lihasheikkoutta); ortopedisten laitteiden käyttäminen kävelyn helpottamiseksi. Ota tarvittaessa särkylääkkeitä tai antikonvulsantteja. [8]
Korostettujen litteiden jalkojen tapauksessa osteotomia voidaan suorittaa, ja kantojen muodonmuutosten yhteydessä on osoitettu niiden kirurginen korjaus - niveltulehdus. [9]
Tutkimusta tehdään sekä taudin geneettisestä komponentista että sen hoitomenetelmistä. Kantasolujen, joidenkin hormonien, lesitiinin tai askorbiinihapon käyttö ei ole vielä tuottanut positiivisia tuloksia.
Mutta viimeisimmän tutkimuksen ansiosta lähitulevaisuudessa Charcot-Marie-Tooth -taudin hoidossa voi todella esiintyä uusi. Joten vuodesta 2014 lähtien ranskalainen yritys Pharnext on kehittänyt ja vuoden 2019 puolivälistä lähtien kliinisiä tutkimuksia lääkkeestä PXT3003 CMT-tyypin 1 hoitamiseksi aikuisilla, tukahduttamalla PMP22-geenin lisääntynyttä ilmentymistä, parantamalla ääreishermojen myelinaatiota ja heikentävät hermo-lihasoireet.
Lääketieteellisen yhtiön Sarepta Therapeutics (USA) asiantuntijat työskentelevät tyypin 1 Charcot-Marie-Tooth -taudin geeniterapian parissa. Tässä terapiassa käytetään Dependovirus-suvun vaarattomaa adeno-assosioitunutta virusta (AAV), jossa on lineaarinen yksisäikeinen DNA-genomi, joka kuljettaa kehoon NTF3-geenin, joka koodaa neurotrofiini-3 (NT-3) -proteiinia. Schwannin hermosolujen toimintaa.
Vuoden 2020 loppuun mennessä Helixmith aloittaa Etelä-Koreassa kehitetyn Engensis-geeniterapian (VM202) kliiniset tutkimukset lihasten oireiden hoitamiseksi tyypin 1 CMT: ssä. [10]
Ennaltaehkäisy
CMT: n ehkäisy voi olla tulevien vanhempien geneettistä neuvontaa, varsinkin jos jollakin avioparilla on tämä tauti perheessä. De novo -geenipistemutaatioiden tapauksia on kuitenkin tunnistettu, toisin sanoen taudin puuttuessa suvussa.
Raskauden aikana kuorionvillanäytteenotto (10–13 raskausviikkoa) sekä lapsiveden analyysi (15–18 viikon kohdalla) mahdollistavat Charcot-Marie-Tooth-taudin todennäköisyyden tarkastamisen syntymättömällä lapsella.
Ennuste
Yleensä erityyppisten Charcot-Marie-Tooth-taudin ennuste riippuu kliinisestä vakavuudesta, mutta joka tapauksessa tauti etenee hitaasti. Monilla potilailla on vamma, vaikka tämä ei lyhennä elinajanodotetta.