Lääketieteen asiantuntija

Uudet julkaisut
Charcot-Marie-Toothin tauti.
Viimeksi tarkistettu: 12.07.2025

Kaikki iLive-sisältö tarkistetaan lääketieteellisesti tai se tarkistetaan tosiasiallisen tarkkuuden varmistamiseksi.
Meillä on tiukat hankintaohjeet ja vain linkki hyvämaineisiin mediasivustoihin, akateemisiin tutkimuslaitoksiin ja mahdollisuuksien mukaan lääketieteellisesti vertaisarvioituihin tutkimuksiin. Huomaa, että suluissa ([1], [2] jne.) Olevat numerot ovat napsautettavia linkkejä näihin tutkimuksiin.
Jos sinusta tuntuu, että jokin sisältö on virheellinen, vanhentunut tai muuten kyseenalainen, valitse se ja paina Ctrl + Enter.
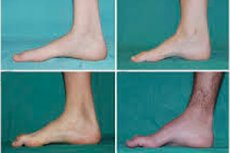
Peroneaalinen lihasatrofia, Charcot-Marie-Toothin oireyhtymä tai sairaus, on joukko kroonisia perinnöllisiä sairauksia, joissa vaurioitetaan ääreishermoja.
ICD-10-luokituksen mukaan hermoston sairauksia käsittelevässä osiossa tämän sairauden koodi on G60.0 (perinnöllinen motorinen ja sensorinen neuropatia). Se sisältyy myös harvinaisten sairauksien luetteloon.
Epidemiologia
Kliinisten tilastojen mukaan kaikenlaisten Charcot-Marie-Tooth-taudin esiintyvyys 100 000 asukasta kohden on 19 tapausta (muiden lähteiden mukaan yksi tapaus 2,5–10 000 asukasta kohden).
CMT tyyppi 1 muodostaa noin kaksi kolmasosaa tapauksista (yksi tapaus 5 000–7 000 asukasta kohden), ja lähes 70 % niistä liittyy PMP22-geenin kahdentumiseen. Yli 1,2 miljoonaa ihmistä maailmanlaajuisesti kärsii tästä sairaudesta.
CMT-tyypin 4 ilmaantuvuudeksi arvioidaan 1–5 tapausta 10 000 lasta kohden. [ 1 ]
Syyt Charcot-Marie-Toothin tauti
Polyneuropatiaoireyhtymien luokituksen mukaan peroneaalinen (fibulaarinen) lihasatrofia, Charcot-Marie-Toothin hermoamyotrofia tai Charcot-Marie-Toothin tauti (lyhennettynä CMT) viittaa geneettisesti määräytyviin motoris-sensorisiin polyneuropatioihin. [ 2 ]
Eli sen esiintymisen syyt ovat geneettiset mutaatiot. Ja geneettisten poikkeavuuksien luonteesta riippuen erotetaan tämän oireyhtymän päätyypit tai lajit: demyelinoiva ja aksonaalinen. Ensimmäiseen ryhmään kuuluu Charcot-Marie-Toothin tauti tyyppi 1 (CMT1), joka johtuu PMP22-geenin kahdentumisesta kromosomissa 17, joka koodaa transmembraanista perifeeristä myeliiniproteiinia 22. Tämän seurauksena aksonitupen (hermosoluprosessien) segmentaalinen demyelinaatio ja hermosignaalin johtumisen nopeuden hidastuminen. Lisäksi mutaatioita voi olla joissakin muissa geeneissä.
Aksonaalinen muoto on Charcot-Marie-Toothin tauti tyyppi 2 (CMT2), joka vaikuttaa itse aksoneihin ja liittyy patologisiin muutoksiin MFN2-geenissä lokuksessa 1p36.22. Tämä geeni koodaa kalvoproteiinia mitofusiini-2, joka on välttämätön mitokondrioiden fuusiolle ja toiminnallisten mitokondrioverkostojen muodostumiselle ääreishermosoluissa. CMT2:sta on yli tusina alatyyppiä (joilla on mutaatioita tietyissä geeneissä).
On huomattava, että tällä hetkellä on tunnistettu yli sata geeniä, joiden periytyvä vaurio aiheuttaa Charcot-Marie-Toothin taudin eri alatyyppejä. Esimerkiksi RAB7-geenin mutaatiot johtavat CMT-tyyppiin 2B; SH3TC2-geenin (joka koodaa yhtä Schwannin solujen kalvoproteiineista) muutos aiheuttaa CMT-tyyppiin 4C, joka ilmenee lapsuudessa ja jolle on ominaista motoristen ja sensoristen hermosolujen demyelinaatio (tämän taudin tyyppiä 4 on tusina ja puolitoista muotoa).
Harvinainen tyypin 3 CMT (kutsutaan Dejerine-Sottasin oireyhtymäksi) alkaa kehittyä varhaislapsuudessa ja sen aiheuttavat mutaatiot PMP22-, MPZ-, EGR2- ja muissa geeneissä.
Kun CMT-tyyppi 5 esiintyy 5–12-vuotiaana, havaitaan paitsi motorista neuropatiaa (alaraajojen spastisen parapareesin muodossa), myös näkö- ja kuulohermojen vaurioita.
Lihasheikkous ja näköhermon surkastuminen (näön heikkenemisen kera) sekä tasapainovaikeudet ovat tyypillisiä tyypin 6 Charcot-Marie-Toothin taudille. Tyypin 7 Charcot-Marie-Toothin taudissa ei ole pelkästään motoris-sensorista neuropatiaa, vaan myös verkkokalvon sairaus retinitis pigmentosan muodossa.
Miehillä yleisempi X-kromosomiin kytkeytynyt CMT eli Charcot-Marie-Toothin tauti, johon liittyy raajojen tetrapareesi (sekä käsien että jalkojen liikeheikkous), on demyelinoiva tyyppi, ja sen uskotaan johtuvan X-kromosomin pitkän haaran GJB1-geenin mutaatiosta. GJB1-geeni koodaa konneksiini 32:ta, Schwannin solujen ja oligodendrosyyttien transmembraanista proteiinia, joka säätelee hermosignaalien siirtymistä. [ 3 ]
Riskitekijät
CMT:n tärkein riskitekijä on taudin esiintyminen suvussa eli lähisukulaisilla.
Geneetikkojen mukaan, jos molemmat vanhemmat kantavat autosomaalisesti resessiivisesti periytyvää Charcot-Marie-Tooth-taudin geeniä, riski saada lapsi, jolle kehittyy tämä sairaus, on 25 %. Ja riski sille, että lapsi on tämän geenin kantaja (mutta hänellä ei ole oireita), on arviolta 50 %.
X-kromosomiin kytkeytyneessä periytymisessä (kun mutatoitunut geeni on naisen X-kromosomissa) on 50 %:n riski, että äiti siirtää geenin pojalleen, joka kehittää CMT:n. Tauti ei välttämättä ilmene tyttölapsen syntyessä, mutta tyttären pojat (lapsenlapset) voivat periä viallisen geenin, ja tauti kehittyy.
Synnyssä
Kaikissa Charcot-Marie-Tooth-taudin muodoissa sen patogeneesi johtuu perifeeristen hermojen perinnöllisestä poikkeavuudesta: motorisista (liike) ja aistinvaraisista (herkistä).
Jos CMT-tyyppi on demyelinoiva, niin ääreishermojen aksoneja suojaavan myeliinitupen tuhoutuminen tai vika johtaa hermoimpulssien siirron hidastumiseen ääreishermostossa - aivojen, lihasten ja aistinelinten välillä.
Taudin aksonaalisessa tyypissä itse aksonit vaurioituvat, mikä vaikuttaa negatiivisesti hermosignaalien voimaan, mikä ei riitä lihasten ja aistinelinten täydelliseen stimulaatioon.
Lue myös:
Miten Charcot-Marie-Toothin oireyhtymä tarttuu? Vialliset geenit voivat periytyä autosomaalisesti dominanttisti tai autosomaalisesti peittyvästi.
Yleisin tyyppi, autosomaalinen dominantti periytyminen, esiintyy, kun mutatoituneesta geenistä on yksi kopio (jonka toinen vanhemmista kantaa). Ja CMT:n siirtymisen todennäköisyydeksi jokaiselle syntyneelle lapselle arvioidaan 50 % [ 4 ].
Autosomaalisesti peittyvästi periytyvässä sairaudessa tarvitaan kaksi kopiota viallisesta geenistä (yksi kummaltakin vanhemmalta, jolla ei ole taudin oireita), jotta sairaus voi kehittyä.
40–50 %:ssa tapauksista esiintyy autosomaalisesti dominanttia perinnöllistä demyelinaatiota eli CMT-tyyppiä 1; 12–26 %:ssa tapauksista aksonaalista CMT-tyyppiä 2. Ja 10–15 %:ssa tapauksista havaitaan X-kromosomiin kytkeytynyttä periytymistä. [ 5 ]
Oireet Charcot-Marie-Toothin tauti
Tyypillisesti tämän taudin ensimmäiset oireet alkavat ilmetä lapsuudessa ja nuoruudessa ja kehittyvät vähitellen läpi elämän, vaikka oireyhtymä voi ilmaantua myöhemminkin. Oireiden yhdistelmä on vaihteleva, eikä taudin etenemisnopeutta eikä sen vakavuutta voida ennustaa.
Tyypillisiä alkuvaiheen oireita ovat lisääntynyt yleinen väsymys; jalkojen, nilkkojen ja säärien lihasten heikentynyt sävy (heikkous); refleksien puute. Tämä vaikeuttaa jalkojen liikkumista ja johtaa dysbasiaan (kävelyhäiriöön), joka ilmenee jalkojen korkeampana nousuna, usein toistuvien kompastelujen ja kaatumisten muodossa. Charcot-Marie-Tooth-taudin oireita pienellä lapsella voivat olla huomattava kömpelyys ja ikään liittyvät epätyypilliset kävelyvaikeudet, jotka liittyvät molemminpuoliseen jalkaterän roikkumiseen. Myös jalan epämuodostumat ovat tyypillisiä: korkea jalkaholvi (ontto jalka) tai vaikeat lättäjalat, käyrät (vasaramaiset) varpaat.
Jos lapsella kävellään varpaillaan lihashypotonian taustalla, neurologi voi epäillä tyypin 4 CMT:tä, jossa lapset eivät välttämättä pysty kävelemään murrosiässä.
Taudin edetessä lihasatrofia ja -heikkous leviävät yläraajoihin, mikä vaikeuttaa hienomotoristen taitojen ja normaalien käsien toimintojen suorittamista. Tuntoaistin ja lämmön ja kylmän aistimisen heikkeneminen sekä jalkojen ja käsien tunnottomuus viittaavat tuntohermojen aksonien vaurioitumiseen.
Lapsuusiässä ilmenevässä Charcot-Marie-Toothin taudin tyypeissä 3 ja 6 havaitaan sensorista ataksiaa (liikkeiden ja tasapainon koordinaation heikkenemistä), lihasten nykimistä ja vapinaa, kasvohermovaurioita, näköhermon surkastumista ja nystagmusta sekä kuulon heikkenemistä.
Myöhemmissä vaiheissa voi esiintyä hallitsematonta vapinaa (tremor) ja usein esiintyviä lihaskramppeja; liikkumisvaikeudet voivat johtaa kivun kehittymiseen: lihas-, nivel- ja neuropaattiseen kipuun.
Komplikaatiot ja seuraukset
Charcot-Marie-Toothin taudilla voi olla komplikaatioita ja seurauksia, kuten:
- useammin esiintyviä nyrjähdyksiä ja murtumia;
- nivelten ympärillä olevien lihasten ja jänteiden lyhenemiseen liittyvät kontraktuurat;
- skolioosi (selkärangan kaarevuus);
- hengitysvaikeudet – kun pallean lihaksia hermottavat hermokuidut vaurioituvat:
- kyvyn menetys liikkua itsenäisesti.
Diagnostiikka Charcot-Marie-Toothin tauti
Diagnoosiin kuuluu kliininen tutkimus, anamneesi (mukaan lukien sukututkimus), neurologinen tutkimus ja systeeminen tutkimus.
Testeillä tarkistetaan liikerata, herkkyys ja jännerefleksit. Hermojen johtumista voidaan arvioida instrumentaalisilla diagnostiikoilla – elektromyografialla tai elektroneuromyografialla. Myös ultraääni- tai magneettikuvaus voi olla tarpeen. [ 6 ]
Yleisimpien CMT:tä aiheuttavien geneettisten mutaatioiden havaitsemiseen verinäytteessä käytettävät geeni- tai DNA-testit ovat rajalliset, koska DNA-testejä ei tällä hetkellä ole saatavilla kaikentyyppisille taudeille. Lisätietoja on kohdassa Geneettinen testaus.
Joissakin tapauksissa suoritetaan perifeerisen hermon (yleensä suraalisen hermon) biopsia.
Differentiaalinen diagnoosi
Erotusdiagnoosiin kuuluvat muut perifeeriset neuropatiat, Duchennen lihasdystrofia, myelopatiset ja myasteeniset oireyhtymät, diabeettinen neuropatia, multippeli- ja amyotrofisessa lateraaliskleroosissa esiintyvät myelopatiat, Guillain-Barrén oireyhtymä, peroneaalihermon trauma ja sen surkastuminen (mukaan lukien puristuminen selkärangan lannevälilevyjen väliin), pikkuaivojen tai talamuksen vauriot sekä kemoterapian sivuvaikutukset (sytostaattihoidon, kuten vinkristiinin tai paklitakselin, aikana). [ 7 ]
Kuka ottaa yhteyttä?
Hoito Charcot-Marie-Toothin tauti
Nykyään tämän perinnöllisen sairauden hoitoon kuuluvat liikuntaterapia (joka pyrkii vahvistamaan ja venyttämään lihaksia); toimintaterapia (joka auttaa käsien lihasheikkoudesta kärsiviä potilaita); ja ortopedisten laitteiden käyttö kävelyn helpottamiseksi. Tarvittaessa määrätään kipulääkkeitä tai kouristuslääkkeitä. [ 8 ]
Vaikeissa lattajalkaisuuden tapauksissa voidaan suorittaa osteotomia, ja kantapään epämuodostuman tapauksessa kirurginen korjaus – artrodeesi – on aiheellista. [ 9 ]
Sekä taudin geneettistä komponenttia että sen hoitomenetelmiä tutkitaan parhaillaan. Kantasolujen, tiettyjen hormonien, lesitiinin tai askorbiinihapon käyttö ei ole vielä tuottanut positiivisia tuloksia.
Mutta viimeaikaisen tutkimuksen ansiosta Charcot-Marie-Toothin taudin hoidossa saattaa todellakin ilmetä jotain uutta lähitulevaisuudessa. Niinpä ranskalainen Pharnext-yritys on vuodesta 2014 lähtien kehittänyt ja vuoden 2019 puolivälistä lähtien on ollut käynnissä kliinisiä tutkimuksia PXT3003-lääkkeelle aikuisten CMT tyypin 1 hoitoon, joka estää PMP22-geenin lisääntynyttä ilmentymistä, parantaa ääreishermojen myelinaatiota ja lievittää neuromuskulaarisia oireita.
Lääketieteellisen yrityksen Sarepta Therapeutics (USA) asiantuntijat työskentelevät geeniterapian parissa tyypin 1 Charcot-Marie-Tooth-taudin hoitoon. Tässä terapiassa käytetään vaaratonta Dependovirus-suvun adenoassosioitunutta virusta (AAV), jolla on lineaarinen yksijuosteinen DNA-genomi. Se siirtää elimistöön NTF3-geenin, joka koodaa Schwannin hermosolujen toiminnalle välttämätöntä neurotrofiini-3 (NT-3) -proteiinia.
Helixmith aloittaa eteläkorealaisen Engensis (VM202) -geeniterapian kliiniset tutkimukset vuoden 2020 loppuun mennessä. Tutkimukset on tarkoitettu hoitamaan tyypin 1 CMT:n lihasoireita. [ 10 ]
Ennaltaehkäisy
KMT:n ehkäisyyn voidaan käyttää tulevien vanhempien geneettistä neuvontaa, erityisesti jos jollakulla pariskunnasta on taudin esiintyminen suvussa. On kuitenkin tunnistettu tapauksia, joissa on tapahtunut de novo -pistegeenimutaatioita eli tautia ei ole esiintynyt suvussa.
Raskauden aikana Charcot-Marie-Tooth-taudin todennäköisyyttä syntymättömällä lapsella voidaan tarkistaa istukkavillusbiopsialla (10–13 raskausviikkoa) sekä lapsivesianalyysillä (15–18 viikkoa).
Ennuste
Yleisesti ottaen erityyppisten Charcot-Marie-Tooth-taudin ennuste riippuu kliinisestä vakavuudesta, mutta kaikissa tapauksissa tauti etenee hitaasti. Monilla potilailla on vammoja, vaikka tämä ei lyhennä elinajanodotetta.