Lääketieteen asiantuntija

Uudet julkaisut
Xeroderma pigmentosum
Viimeksi tarkistettu: 04.07.2025

Kaikki iLive-sisältö tarkistetaan lääketieteellisesti tai se tarkistetaan tosiasiallisen tarkkuuden varmistamiseksi.
Meillä on tiukat hankintaohjeet ja vain linkki hyvämaineisiin mediasivustoihin, akateemisiin tutkimuslaitoksiin ja mahdollisuuksien mukaan lääketieteellisesti vertaisarvioituihin tutkimuksiin. Huomaa, että suluissa ([1], [2] jne.) Olevat numerot ovat napsautettavia linkkejä näihin tutkimuksiin.
Jos sinusta tuntuu, että jokin sisältö on virheellinen, vanhentunut tai muuten kyseenalainen, valitse se ja paina Ctrl + Enter.
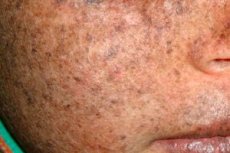
Xeroderma pigmentosum on autosomaalinen peittyvästi periytyvä DNA:n korjaushäiriö. Tauti johtuu mutaatioista geeneissä, jotka osallistuvat ultraviolettisäteilyn ja muiden myrkyllisten aineiden vaurioittaman DNA:n korjaamiseen.
Useimmiten tämä sairaus vaikuttaa lapsiin, joita kutsutaan myös "yön lapsiksi". Taudin yleisiä komplikaatioita ovat tyvisolusyöpä ja muut ihon pahanlaatuiset kasvaimet, metastaattinen pahanlaatuinen melanooma ja okasolusyöpä.
 [ 1 ]
[ 1 ]
Syyt xeroderma pigmentosum
Xeroderma pigmentosum on perinnöllinen sairaus, joka siirtyy autosomaalisen geenin välityksellä vanhemmilta ja sukulaisilta ja jolla on perinnöllinen luonne. Lunch (1967) löysi eräässä seitsemän veljeksen ja sisaren perheessä kliinisiä xeroderma pigmentosumin oireita viidellä heistä.
Synnyssä
UV-endonukleaasientsyymien puutos potilaan soluissa (fibroblasteissa) tai niiden täydellinen puuttuminen on tärkeässä roolissa taudin kehittymisessä. Tämä entsyymi vastaa ultraviolettisäteiden vaurioittaman DNA:n lisääntymisestä. Muiden lähteiden mukaan DNA-polymeraasi-1-entsyymin puutos on tärkeässä roolissa taudin kehittymisessä joillakin potilailla. Tauti kehittyy useimmiten 280–310 nm:n aallonpituisten säteiden vaikutuksesta. Uskotaan, että pigmenttikseroderma kehittyy erilaisten fotodynaamisten aineiden pääsyn seurauksena elimistöön tai porfyriinien lisääntymisen seurauksena ihmisen biologisessa ympäristössä.
Taudin alkuvaiheessa havaitaan hyperkeratoosia, epidermiksen fokuksen surkastumista, Malpighin kerroksen ohenemista, melaniinirakeiden lisääntymistä solujen tyvikerroksessa ja kroonista tulehdusinfiltraattia dermiksen yläkerroksessa (pääasiassa verisuonten ympärillä). Myöhemmin havaitaan degeneratiivisia muutoksia kollageenissa ja elastisissa kuiduissa, ja kasvainvaiheessa paljastuvat ihosyövälle tyypilliset merkit.
Oireet xeroderma pigmentosum
Tämä sairaus vaikuttaa sekä miehiin että naisiin yhtä lailla. Tauti alkaa varhaislapsuudessa keväällä tai kesällä. Potilaan iho on erityisen herkkä auringonvalolle. Siksi ensimmäinen ihottuma, luomen kaltainen hyperpigmentaatio, ilmestyy auringonvalolle altistuville ihoalueille. Ihottuma lisääntyy, punoitus voimistuu, muuttuu tummanruskeaksi, ja siihen ilmestyy telangiektasiaa, angioomaa ja surkastumista. Myöhemmin kasvavat papilloomat ja syyliä (pääasiassa kasvojen ja kaulan iholle), ja näppylöitä ja haavaumia ilmaantuu. Haavaumien arpeutumisen seurauksena nenä ohenee ("linnun nokka") ja silmäluomi kääntyy ulospäin. Papilloomat kehittyvät yleensä pahanlaatuisiksi kasvaimiksi. Pigmenttinen kseroderma esiintyy yleensä yhdessä selkäydinsyövän, melanosarkoomien, kanssa.
Mikä häiritsee sinua?
Vaiheet
Xeroderma pigmentosumin kliinisessä kulussa erotetaan viisi vaihetta: tulehduksellinen (erythematoottinen), hyperpigmentaatio, surkastuminen, hyperkeratoosi ja pahanlaatuiset kasvaimet.
Tulehdusvaiheessa (eryteemavaiheessa) auringonvalolle altistuville ihoalueille (kasvot, kaula, ylärinta, käsivarret, kädet) ilmestyy turvotusta, punaisia läiskiä ja joskus rakkuloita ja vesikkeleitä. Auringonvalolle altistumattomilla alueilla ihottumaa ei juurikaan esiinny.
Hyperpigmentaatiossa punaisten täplien tilalle ilmestyy vaaleanruskeita, ruskeita, luomien kaltaisia hyperpigmentoituneita täpliä.
Surkastumisessa iho kuivuu, ohenee ja rypistyy. Huulten ja nenän iholla havaitaan lukuisia pieniä, tähtimäisiä telangiektasioita ja kiiltäväpintaisia arpia. Surkastumisen ja arpien seurauksena kehittyy mikrotomia (suuaukon pieneneminen), ektropium, korvien ja nenän oheneminen sekä nenäaukon ja suun atresia. 80–85 %:lla potilaista silmät vaurioituvat: havaitaan sidekalvotulehdusta, keratiittia, sarveiskalvon ja limakalvojen vaurioita sekä näön heikkenemistä. Silmäluomien iholla havaitaan dyskromiaa, telangiektasioita, hyperkeratoottisia muutoksia ja kasvaimia.
Hyperkeratoosissa edellä kuvatuissa patologisissa fokaaleissa esiintyy syyläkasvaimia, papilloomaa, keratoakantoomaa, fibroomaa, angiofibrioomaa ja muita hyvänlaatuisia kasvaimia. Siksi jotkut tutkijat sisällyttävät pigmenttikseroderman syöpää edeltävien sairauksien ryhmään.
10–15 vuoden kuluttua taudin puhkeamisesta pahanlaatuisia ihokasvaimia (basaliooma, endoteliooma, angiosarkooma) esiintyy atrofisesti muuttuneissa pesäkkeissä ja pigmenttiläiskissä. Kasvaimet tuhoutuvat lyhyessä ajassa, lähettävät etäpesäkkeitä sisäelimiin ja johtavat kuolemaan. Joillakin potilailla sisäelimissä ja kudoksissa havaitaan yleisiä dystrofisia muutoksia (toisen ja kolmannen varpaan syndaktelia, hammasdystrofia, täydellinen hiustenlähtö jne.).
Lomakkeet
Xeroderma pigmentosumin neurologinen muoto ilmenee kahdessa oireyhtymässä.
Reedin oireyhtymälle on ominaista kseroderma pigmentosumin ja mikrokefalian kliiniset ilmentymät, idiopatia ja luuston hidas kasvu. Taudin neurologisessa muodossa DNA:n korjaus on vaikeaa ja röntgenhoito johtaa tärkeimpien patologisten pesäkkeiden lisääntymiseen.
De Sinctis-X Cocchione -oireyhtymälle on ominaista seuraavat kliiniset oireet:
- xeroderma pigmentosumin kliinisten oireiden kehittyminen erityisen valoherkälle iholle;
- pahanlaatuisten kasvainten varhainen esiintyminen;
- samanaikainen spastisen halvauksen, mikrokefalian ja dementian kulku;
- synnynnäiset epämuodostumat ja lyhytkasvuisuus;
- sukurauhasten alikehittyminen;
- usein keskenmenoja;
- resessiivinen periytyminen perinnöllisesti.
Joidenkin ihotautilääkäreiden mukaan Santis-Cacchione-oireyhtymä ei ole itsenäinen sairaus, vaan vakava ja täysin ilmaistu kliininen ilmentymä xeroderma pigmentosumista.
Geneettiset muodot
Tyypit |
Geeni |
Lokus |
Kuvaus |
Tyyppi A, I, XPA |
XPA |
9q22.3 |
Xeroderma pigmentosum (XP) ryhmä A – klassinen muoto |
Tyyppi B, II, XPB |
XPB |
2q21 |
XP-ryhmä B |
Tyyppi C, III, XPC |
XPC |
3p25 |
XP-ryhmä C |
Tyyppi D, IV, XPD |
XPD ERCC6 |
19q13.2–q13.3, 10q11 |
XP Group D tai De Sanctis-Cacchione -oireyhtymä |
Tyyppi E, V, XPE |
DDB2 |
11s.12-s.11 |
XP-ryhmä E |
Tyyppi F, VI, XPF |
ERCC4 |
16s.13.3–13.13 |
XP-ryhmä F |
Tyyppi G, VII, XPG |
RAD2ERCC5 |
13q33 |
XP-ryhmä G ja COFS-oireyhtymä (cerebro-oculo-facioskeletal syndrome) tyyppi 3 |
Tyyppi V, XPV |
POLH |
6s. 21.1–12 |
Xeroderma Pigmentosa -variantti |
Mitä on tutkittava?
Kuinka tarkastella?
Kuka ottaa yhteyttä?
Hoito xeroderma pigmentosum
Malarialääkkeet (delagyl, plaquenil, resoquin jne.), jotka suojaavat DNA:ta ultraviolettisäteiltä, estävät depolymeroitumisprosessia ja vähentävät ihon herkkyyttä (valoherkkyyttä) auringonvalolle. Näillä lääkkeillä on tulehdusta estäviä ja herkistäviä ominaisuuksia. Yleishoitoa suositellaan yhdessä vitamiinihoidon (B1, B2, PP, B6, B12, A, E), antihistamiinien (tavegil, difenhydramiini, suprastin) ja herkistävien aineiden (natriumtiosulfaatti, 10 % kalsiumkloridia laskimoon 10 ml) kanssa.
Paikalliseen hoitoon käytetään aurinkosuojavoiteita ja -voiteita.
Pigmenttikseroderman kasvainmuodossa käytetään kirurgisia menetelmiä, nestemäistä typpeä ja lasersäteitä. Suojautuaksesi auringonvalolta sinun on käytettävä löysiä vaatteita, aurinkohattuja ja käsineitä.
Ennuste
Useimmat potilaat (2/3) kuolevat ennen 15 vuoden ikää. Jotkut potilaat voivat elää 40–50-vuotiaaksi.
[ 20 ]