Lääketieteen asiantuntija

Uudet julkaisut
Amauroottinen idiotismi
Viimeksi tarkistettu: 04.07.2025

Kaikki iLive-sisältö tarkistetaan lääketieteellisesti tai se tarkistetaan tosiasiallisen tarkkuuden varmistamiseksi.
Meillä on tiukat hankintaohjeet ja vain linkki hyvämaineisiin mediasivustoihin, akateemisiin tutkimuslaitoksiin ja mahdollisuuksien mukaan lääketieteellisesti vertaisarvioituihin tutkimuksiin. Huomaa, että suluissa ([1], [2] jne.) Olevat numerot ovat napsautettavia linkkejä näihin tutkimuksiin.
Jos sinusta tuntuu, että jokin sisältö on virheellinen, vanhentunut tai muuten kyseenalainen, valitse se ja paina Ctrl + Enter.

Amauroottinen idiotismia on harvinainen etenevä sairaus. Sille on ominaista näön asteittainen heikkeneminen täydelliseen sokeuteen ja älykkyyden heikkeneminen, kunnes idiotismia ilmenee. Tämän seurauksena potilaalle kehittyy syvä marasmus, joka johtaa kuolemaan. Silmälääkäri Dr. Tau kuvasi taudin ensimmäisen kerran yli 130 vuotta sitten. Tau pani merkille silmänpohjan erityisen muutoksen. Tautia on jo kuvattu yli 500 tapausta.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ]
Epidemiologia
Taudin epidemiologiassa on todettu perinnöllinen luonne. Perheissä, joissa on amauroottista idioottisuutta sairastavia potilaita, verisukulaisuusliitot ovat vaarallisia samasta syystä. Myös kanadalais-ranskalaista tai juutalaista alkuperää olevat ihmiset ovat vaarassa.
[ 11 ], [ 12 ], [ 13 ], [ 14 ], [ 15 ], [ 16 ], [ 17 ], [ 18 ]
Syyt amauroottinen idiotismi
Huolimatta lukuisista taudista kerätyistä tiedoista, tiedemiehet etsivät edelleen vastauksia moniin kysymyksiin amauroottisen idiotismin syistä, patogeneesistä ja jopa ilmentymistä.
On esitetty, että tauti on perinnöllinen. Perintötyyppi on autosomaalisesti peittyvä. Useimmiten sairaus vaikuttaa aivopuoliskojen pikkuaivoihin ja takaraivolohkoihin, ja sillä on vakavia seurauksia ja komplikaatioita koko keholle: näköhermojen surkastumista, hermokuidut voivat menettää kalvonsa ja hermosolujen väliset yhteydet voivat hajota.
Useimmat asiantuntijat myöntävät, että taudin kliiniset oireet voivat olla melko vaihtelevia ja korreloida sen iän kanssa, jolloin amauroottinen idioottisuus alkoi kehittyä potilaalla.
Taudin syitä tutkittaessa havaittiin tietty kaava: tauti vaikuttaa usein saman perheen lapsiin, minkä vuoksi käytetään nimitystä "perheellinen amauroottinen idiotismi". Tutkimusten mukaan, joiden tulokset julkaistiin taudin tutkimuksen vasta alussa, 64:stä amauroottisen idiotismin tapauksesta 37 todettiin 13 perheessä (jokaisessa perheessä oli 2-5 sairasta lasta). On huomionarvoista, että tällaisissa perheissä sairailla oli täysin terveitä veljiä ja sisaria. Nykyään uskotaan, että resessiivisellä periytymisellä on suuri rooli taudin kehittymisessä. Siten on mahdollista selittää taudin esiintymistiheys samoissa perheissä. Analysoitaessa perinnöllisyystekijää amauroottisen idiotismin syynä, ei pidä rajoittua kliinisesti ilmenevien oireiden esiintymiseen potilasperheissä (sekä nousevassa että lateraalisessa linjassa), vaan on otettava huomioon myös alkeelliset oireet, esimerkiksi näkölaitteen toiminnan ominaispiirteet (familiaalinen suonikalvontulehdus, pigmenttinen verkkokalvon dystrofia jne.).
Oireet amauroottinen idiotismi
Ensimmäiset synnynnäisen muodon merkit ilmenevät eliniän ensimmäisinä päivinä tai viikkoina. Vauva syntyy vesipään tai mikrokefalian kanssa, kärsii kohtauksista, halvaantumisesta ja hengitysvaikeuksista. Lapsi kuolee muutaman kuukauden kuluttua.
Vaiheet
Infantiili muoto kehittyy 4–6 kuukauden iässä. Tälle amauroottisen idiopatian muodolle on ominaista perinnöllinen luonne. Näkö heikkenee nopeasti: vauva ei pysty kiinnittämään katsettaan eikä havaitse esineitä. Silmänpohjaan ilmestyy niin sanottu "kirsikankuoppa" – punertava täplä makulan alueella, jota ympäröi harmaanvalkoinen reunus. Sitten näköhermot surkastuvat, ja lapsi menettää kokonaan näkökyvyn. Suuntautumiskyky, suojarefleksit ja liikkumiskyky menetetään vähitellen. Potilaat reagoivat voimakkaasti ääniärsykkeisiin – he säpsähtävät äänestä, joka on terveelle ihmiselle hiljainen, ja kouristuksia voi esiintyä lisääntyneen lihasjänteyden vuoksi. Taudin loppuvaiheessa kehittyy yleinen surkastuminen, kehon uupumus ja kaikkien ojentajalihasten lisääntynyt sävy. Myös taudin ennuste on pettymys: potilas kuolee puolitoista–kaksi vuotta taudin alkamisen jälkeen.
Myöhäislapsuusiän muoto alkaa 3–4 vuoden iässä. Etenevä sairaus vuorottelee remissiovaiheiden kanssa. Älykkyyden asteittaiseen menetykseen liittyy kohtauksia, koordinaatiohäiriöitä ja ekstrapyramidaalisia häiriöitä. Tälle muodolle on ominaista myös näköhermon surkastuminen. Kuolema tapahtuu 6–8 vuotta amauroottisen idiotismin alkamisen jälkeen.
Nuoruusiän muoto alkaa ilmetä 6–10 vuoden iässä. Spielmeyerin amauroottinen idiotismia etenee hitaammin. Silmänpohjan muutokset tapahtuvat samanaikaisesti pigmenttimäisen verkkokalvon dystrofian ilmenemismuotojen kanssa. Potilaan näkökyky heikkenee vähitellen, samoin kuin älykkyys. Motoristen toimintojen heikkeneminen voi ilmetä eri tavoin ja epäsäännöllisesti: esiintyy ei kovin voimakasta käsien ja jalkojen halvaantumista, ekstrapyramidaalisia ja bulbaarisia häiriöitä. Tauti johtaa kuolemaan 10–25 vuotta ensimmäisten oireiden kehittymisen jälkeen.
Myöhäinen muoto esiintyy hyvin harvoin ja kehittyy äärimmäisen hitaasti. Potilaan mielentila muuttuu (kuten orgaaninen mielenterveysoireyhtymä), havaitaan näköhermojen surkastumista ja verkkokalvon pigmenttidystrofiaa. Viimeiselle vaiheelle on ominaista halvaus ja epileptiforminen oireyhtymä. Potilas kuolee 10–15 vuotta taudin alkamisen jälkeen.
[ 38 ], [ 39 ], [ 40 ], [ 41 ], [ 42 ], [ 43 ], [ 44 ], [ 45 ]
Lomakkeet
Amauroottista idioottisuutta on neljää tyyppiä:
- Tay-Sachsin oireyhtymä (ilmenee jo nuorella iällä);
- Jansky-Bilynovsky (esiintyy lapsilla myöhemmällä iällä);
- Spielmeyer-Vogtin oireyhtymä (esiintyy nuorilla);
- Kufsa (myöhäinen muoto).
Jotkut tiedemiehet erottavat myös synnynnäisen Norman-Wood-tyypin erikseen.
Jokaisella tautityypillä on omat kliiniset ilmentymänsä, mutta niitä kaikkia yhdistävät yhteiset syyt, kliininen kuva, anatominen perusta ja patogeneesi.
[ 46 ], [ 47 ], [ 48 ], [ 49 ], [ 50 ], [ 51 ], [ 52 ], [ 53 ], [ 54 ]
Diagnostiikka amauroottinen idiotismi
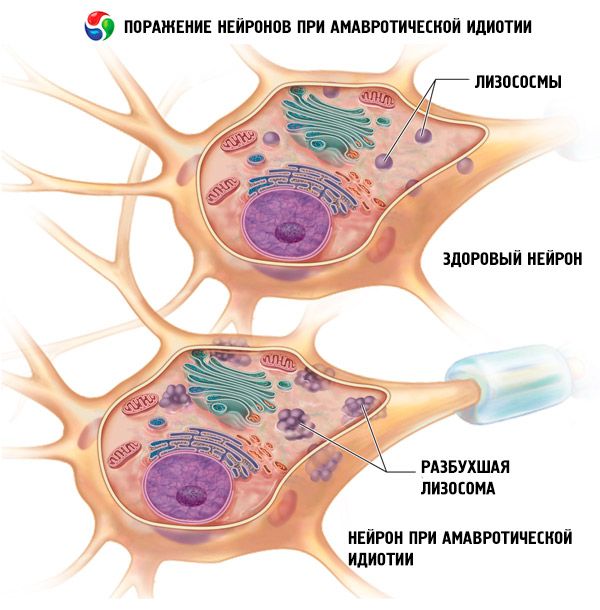
Amauroottinen idioottisuus johtuu lipidiaineenvaihdunnan häiriöstä, jonka seurauksena lipidiaineenvaihdunnan välituote, sfingomyeliini, kertyy kehon eri soluihin. Kertymien sijainti ja koostumus määräävät taudin spesifisen kliinisen kuvan kehittymisen.
[ 55 ], [ 56 ], [ 57 ], [ 58 ], [ 59 ], [ 60 ], [ 61 ], [ 62 ], [ 63 ]
Kuinka tarkastella?
Differentiaalinen diagnoosi
Amauroottisen idiotismin differentiaalinen diagnoosi perustuu spesifiseen kliiniseen kuvaan ja fundus-tyypillisiin patologioihin.
Varhaisessa muodossa on samanlaisia oireita kuin Landingin taudissa, joka on eräs mukopolysakkaridoosin tyyppi. Landingin tauti kehittyy syntymän jälkeisistä ensimmäisistä kuukausista lähtien ja johtaa kuolemaan 2–3 vuoden kuluttua. Silmänpohjaan ilmestyy "kirsikankuori" 1/5 tapauksista, verkkokalvon degeneratiiviset muutokset ja äänten havaitsemisen vääristyminen (hyperkussio) eivät ole käytännössä tyypillisiä, mutta samanaikaista pernan ja maksan suurenemista, mielenterveyshäiriöitä ja liikehäiriöitä havaitaan.
Nuoruusiän muoto esiintyy joskus samoissa oireissa kuin Lawrence-Moon-Biedlin oireyhtymä. Näiden sairauksien erottamiseksi on kiinnitettävä huomiota niiden muihin ilmenemismuotoihin. Lawrence-Moon-Biedlin oireyhtymälle on ominaista nopea painonnousu, raajojen epämuodostuma, jolle on ominaista lisäsormien tai -varpaiden esiintyminen, havaittavat vegetatiiviset ja troofiset häiriöt sekä motoristen toimintojen häiriöiden puuttuminen.
Myöhäisen amauroottisen idiotismin oireiden monimuotoisuus vaikeuttaa diagnoosin tekemistä elämän aikana. Sen ilmenemismuodot ovat samankaltaisia kuin Friedreichin ataksia, multippeliskleroosi, Alzheimerin tauti, Pickin tauti, etenevä halvaus ja jopa skitsofrenia.
Jotkut kirjoittajat väittävät, että tämän taudin diagnoosi, erityisesti silloin, kun kliiniset oireet ovat epäselviä, voidaan luotettavasti todeta vasta kuoleman jälkeen hermoston histologisten poikkeavuuksien analyysin perusteella.
Kuka ottaa yhteyttä?
Hoito amauroottinen idiotismi
Ei ole olemassa järkevää ja tehokasta hoitoa. Nykyään amauroottisen idiotismin hoito on suunnattu yksinomaan oireiden lievittämiseen. Käytetään rauhoittavia lääkkeitä, nootrooppisia lääkkeitä, kouristuslääkkeitä ja yleisiä tonikuja.
Aivojen verenkierron ja aineenvaihduntaprosessien aktivoimiseksi määrätään glysiiniä, elkaria, cerebrolysiinia, glutamiinihappoa ja pantogaamia.
Kouristusoireyhtymän lievittämiseksi määrätään difeniinia tai karmatsepiinia.
Positiivinen tulos voidaan saavuttaa käyttämällä kudosuutteita, verensiirtoa tai plasmaa.
Ennaltaehkäisy
Tehokkaan amauroottisen idiotismin hoidon puute pakottaa meidät kiinnittämään erityistä huomiota ennaltaehkäisyyn. On jo olemassa menetelmiä, joilla voidaan tunnistaa patologisen geenin heterotsygoottiset kantajat, ja menetelmiä amauroottisen idiotismin diagnosoimiseksi raskauden aikana. Taudin synnytystä edeltävässä diagnostiikassa analysoidaan heksosaminidaasi A:n aktiivisuutta lapsivedessä. Jos havaitaan vähentynyttä entsyymiaktiivisuutta, suositellaan raskauden keskeyttämistä. Sairaan lapsen vanhempia kehotetaan lopettamaan lasten saaminen.
Использованная литература