Lääketieteen asiantuntija

Uudet julkaisut
Alkaptonuria on synnynnäinen entsyymipoikkeavuus, -
Viimeksi tarkistettu: 04.07.2025

Kaikki iLive-sisältö tarkistetaan lääketieteellisesti tai se tarkistetaan tosiasiallisen tarkkuuden varmistamiseksi.
Meillä on tiukat hankintaohjeet ja vain linkki hyvämaineisiin mediasivustoihin, akateemisiin tutkimuslaitoksiin ja mahdollisuuksien mukaan lääketieteellisesti vertaisarvioituihin tutkimuksiin. Huomaa, että suluissa ([1], [2] jne.) Olevat numerot ovat napsautettavia linkkejä näihin tutkimuksiin.
Jos sinusta tuntuu, että jokin sisältö on virheellinen, vanhentunut tai muuten kyseenalainen, valitse se ja paina Ctrl + Enter.
Yksi hyvin harvinaisista aineenvaihduntahäiriöistä, alkaptonuria, viittaa synnynnäisiin poikkeavuuksiin aminohappo tyrosiinin aineenvaihdunnassa.
Tätä oireyhtymää voidaan kutsua myös homogentisaattioksidaasin puutteeksi, homogentisinuriaksi, perinnölliseksi okronoosiksi tai mustavirtsaiseksi sairaudeksi.[ 1 ]
Epidemiologia
Tilastojen mukaan alkaptonuriaa esiintyy enintään yhdeksän miljoonaa ihmistä kohden. Ja useimmissa Euroopan maissa tapauksia on yksi 100–250 tuhatta elävänä syntynyttä lasta kohden.
Euroopan maista poikkeuksena on Slovakia (erityisesti suhteellisen pieni luoteisalue), jossa alkaptonurian esiintyvyys on yksi tapaus 19 000 vastasyntynyttä kohden. Tämä johtuu todennäköisesti siitä, että siellä asuvien slovakialaisten romaniperheiden keskuudessa sisäsiitoksen (serkkujen välisten avioliittojen) taso on Euroopan korkein: 10–14 %. [ 2 ]
Syyt alkaptonuria
Alkaptonurian, aromaattisen (homosyklisen) α-aminohapon tyrosiinin katabolian (aineenvaihdunnan hajoamisen) synnynnäisenä häiriönä, tarkat syyt on selvitetty: tämän tyyppinen aineenvaihduntahäiriö on seurausta homotsygoottisista tai yhdistelmäheterotsygoottisista mutaatioista yhdessä tuhansista kromosomin 3 geeneistä, tarkemmin sanottuna HGD-geenistä, joka sijaitsee kromosomin pitkällä haaralla lokuksessa 3q21-q23. Tämä geeni koodaa maksaentsyymin homogentisaatti-1,2-dioksigenaasin [ 3 ] (jota kutsutaan myös homogentisiinihappooksidaasiksi tai homogentisaattioksidaasiksi) nukleotidisekvenssejä. Tämä entsyymi on rautaa sisältävä metalloproteiini, jota tarvitaan yhteen tyrosiinin hajoamisvaiheista kehossa [ 4 ], [ 5 ].
Alkaptonuria on siis homogentisaatti-1,2-dioksigenaasi-entsyymin vika tai tarkemmin sanottuna sen geneettisesti määräytyneen puutteen tai täydellisen puuttumisen seuraus. [ 6 ]
Koska alkaptonuria on synnynnäinen entsyymipuutos, se periytyy autosomaalisesti peittyvästi. Jotta alkaptonuriaa esiintyisi lapsilla, molemmilla vanhemmilla on oltava modifioitu entsyymigeeni, koska kumpikin vanhemmista siirtää lapselle vain yhden kopion kahdesta saatavilla olevasta geenistä.
Uusimpien tietojen mukaan HGD-geenin modifikaatioita on yli kaksisataa, ja useimmiten havaitaan missense-mutaatioita, translokaatiota ja silmukointia.
Riskitekijät
Ainoa riskitekijä tämän synnynnäisen entsymopatian kehittymiselle on sen esiintyminen suvussa ja kahden muuntuneen HGD-geenin kopion periytyminen, jos vanhemmilla ei esiinny alkaptonuriaa (poikkeavuuden siirtymisriski on 25 %) tai jos toisella vanhemmista on tämä sairaus. [ 7 ]
Synnyssä
Tyrosiinilla on keskeinen rooli proteiinien synteesissä, kromoproteiinien – ihon pigmentti melaniinin – sekä kilpirauhashormonien ja katekoliamiinien välittäjäaineiden tuotannossa.
Solujen tyrosiinin määrän säätelymekanismi on hyvin monimutkainen, ja keho normalisoi ylimääräisen tyrosiinin pitoisuuden hajottamalla sitä. Tyrosiinin kataboliaprosessi, kuten kaikkien aromaattisten aminohappojen, on monivaiheinen ja tapahtuu useissa vaiheissa. Jokainen tyrosiinin metabolisen hajoamisen vaihe tapahtuu tietyn entsyymin osallistuessa ja välituotteen muodostuessa.
Joten ensin aminohappo hajoaa para-hydroksifenyylipyruvaatiksi, joka muuttuu alkaptoniksi – 2,5-dihydroksifenyylietikka- eli homogentisiinihapoksi. Sitten alkaptonin pitäisi muuttua maleetikkahapoksi, mutta näin ei tapahdu. [ 8 ]
Ja alkaptonurian patogeneesi koostuu tyrosiinin katabolian biokemiallisten reaktioiden lopettamisesta homogentisiinihapon muodostumisvaiheessa: sen hajottamiseen ei yksinkertaisesti tarvita entsyymiä – homogentisiinioksidaasia.
Homogentisiinihappoa ei elimistö käytä, ja se voi kertyä munuaisten kautta erittymisen myötä. Lisäksi se hapettuu bentsokinoasetaatiksi (bentsokinonetikkahapoksi), joka sitoutumalla kudosten ja kehon nesteiden molekyyleihin muodostaa melaniinin kaltaisia biopolymeeriyhdisteitä.
Näiden välituotteiden kertyminen kudokseen johtaa rustokudoksen kollageenirakenteen häiriintymiseen, mikä vähentää sen elastisuutta – ja aiheuttaa monia alkaptonurian kliinisiä oireita ja komplikaatioiden kehittymistä.
Oireet alkaptonuria
Vastasyntyneiden ja imeväisten alkaptonurialle on ominaista virtsan tummuminen. Ilman vaikutuksesta vaippoihin, alusvaatteisiin ja housutappeihin kertynyt virtsa muuttuu tummanruskeaksi; tämä johtuu homogentisiinihapon kertymisestä ja vapautumisesta, joka hapettuu bentsokinoasetaatiksi. [ 9 ]
Muiden oireiden puuttuessa alkaptonuriaa pienillä lapsilla ei usein tunnisteta ajoissa, koska virtsa voi tummua useiden tuntien virtsaamisen jälkeen. Joidenkin tietojen mukaan vain viidennes alle 12 kuukauden ikäisistä lapsista, joilla on syntynyt tämä entsyymipuutos, havaitaan kliinisessä ympäristössä. Siksi on erittäin tärkeää, että vanhemmat kiinnittävät huomiota imeväistensä hoitoon.
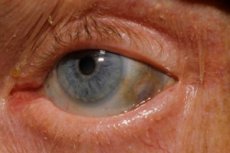
Lisäksi varhaisia oireita ovat silmien kovakalvon sekä korvien ja nenän rustojen pigmentaatio (sinertävän harmaa väri), jota usein kutsutaan okronoosiksi.[ 10 ]
Ajan myötä ilmenee muita oireita:
- ihon voimakas pigmentaatio poskipäissä, kainaloissa ja sukupuolielimissä;
- vaatteiden tahraantuminen kosketuksissa kehon hikoilevien alueiden kanssa;
- yleisen heikkouden hyökkäykset;
- käheä ääni.
On pidettävä mielessä, että alkaptonuria ja okronoosi, kuten edellä todettiin, ovat synonyymejä samalle tyrosiinin katabolian häiriölle.
Vaahterasiirappivirtsasairaus ja alkaptonuria. Synnynnäinen vaahterasiirappivirtsasairaus eli leusinoosi on myös aineenvaihduntahäiriö, jolla on sama periytymismalli, ja jopa mutaatioita esiintyy samassa kromosomissa, mutta ne vaikuttavat haaraketjuisen α-ketohappodehydrogenaasin entsyymikompleksia koodaavaan geeniin. Tämän vuoksi elimistö ei pysty hajottamaan tiettyjä proteiinien komponentteja, erityisesti aminohappoja leusiinia, isoleusiinia ja valiinia. Tässä sairaudessa virtsalla (ja korvavahalla) on makea tuoksu; lisäksi tämän tyyppisen orgaanisen asidemian kliiniseen kuvaan kuuluvat hypopigmentaatio, verenpaineen vaihtelut, kouristukset, oksentelu ja ripuli, verensokeritason lasku, ketoasidoosi, hallusinaatiot jne. Lasten kuolleisuus on melko korkea; aikuisilla ilman hoitoa voi esiintyä kooma ja kuolema aivoödeeman vuoksi.
Albinismia ja alkaptonuriaa "yhdistää" ainoastaan tyrosiini. Albinismi, myös silmän ja ihon välinen albinismi, johtuu geneettisistä mutaatioista, jotka vaikuttavat melaniinipigmentin tuotantoon. Synnynnäisiä muutoksia havaitaan TYR-geenissä kromosomissa 11 (11q14.3), joka koodaa tyrosinaasia, kuparia sisältävää melanosomientsyymiä, jota tarvitaan ihopigmentin muodostumiseen tyrosiinin aineenvaihduntatuotteiden perusteella. Tämä sairaus on paljon yleisempi kuin alkaptonuria.
Komplikaatiot ja seuraukset
Alkaptonurian seuraukset ja komplikaatiot johtuvat tyrosiinin välituotteiden – homogentisiini- ja bentsokinonietikkahappojen – vaikutuksesta reaktiivisten pigmentoituneiden polymeerien kerrostumisesta, kollageenifibrillien tuhoutumisesta ja ruston tilan heikkenemisestä (niiden mekaanisen rasituksen kestävyyden heikkenemisestä).
Vuosien mittaan aikuisuudessa kehittyy suurten nivelten (lonkka, risti- ja iliakaaliluu sekä polvi) degeneratiivista niveltulehdusta ja nivelrikkoa; nikamavälit kapenevat (erityisesti lanne- ja rintarangassa) – kalkkeutumisen ja osteofyyttien muodostumisen myötä; subkondraalisten luulevyjen kudostiheys vähenee ja alla olevat luut voivat muotoutua uudelleen patologisesti, jolloin muodostuu kasvaimia ja muodonmuutoksia. [ 11 ]
Samasta kalkkeutumisesta johtuen voidaan havaita sydänläppien (aortta- ja mitraaliläpän) ja sepelvaltimoiden vaurioita – joihin liittyy sepelvaltimotaudin merkkejä – sekä munuais- ja eturauhaskivien muodostumista. [ 12 ], [ 13 ]
Diagnostiikka alkaptonuria
Tyypillisesti synnynnäisten aineenvaihduntahäiriöiden diagnoosi perustuu kehon biologisten nesteiden tutkimukseen.
Minkä testien ja reaktioiden perusteella alkaptonuria voidaan diagnosoida? Virtsatestit ovat välttämättömiä homogentisiinihapon havaitsemiseksi ja sen pitoisuuden määrittämiseksi (normaali – 20–30 mg päivässä, kohonnut – 3–8 g). Virtsanäyte tutkitaan kaasukromatografialla tai massaspektrometrialla käyttäen nestekromatografiaa; seulontatesti rautakloridin esiintymiseksi virtsassa on mahdollinen. [ 14 ]
On myös menetelmä nopeaan diagnostiikkaan – alkaptonin määrittäminen kuivuneista virtsan tahroista paperilla (värin intensiteetin perusteella).
Diagnoosin selventämisessä instrumentaalinen diagnostiikka (röntgenkuvaus) sisältää nivelrikon ja muiden nivelpatologioiden radiologisten merkkien tunnistamisen potilailla.
Diagnoosi vahvistetaan perinnöllisten sairauksien molekyyligeneettisillä menetelmillä, kuten geenitestillä ja DNA-sekvensoinnilla. [ 15 ]
Differentiaalinen diagnoosi
Differentiaalidiagnoosiin kuuluvat vastasyntyneen hemokromatoosi ja akuutti maksan vajaatoiminta, melaninuria, akuutti ajoittainen porfyria, hemofagosyyttinen lymfohistiosytoosi, primaarinen mitokondriaalinen patologia, nivelreuma, selkärankareuma.
Kuka ottaa yhteyttä?
Hoito alkaptonuria
Alkaptonurian pääasiallinen hoito on suun kautta otettavat suuret askorbiinihappoannokset (vähintään 1000 mg päivässä). Lapsilla tämä lisää homogentisiinihapon erittymistä virtsaan, ja aikuisilla se vähentää sen johdannaisen, bentsokinonietikkahapon, pitoisuutta virtsassa ja hidastaa sen sitoutumista nivelten sidekudosrakenteisiin ja kollageeniin. [ 16 ]
Länsieurooppalaiset klinikat testaavat Nitisinonia (Orfalin), metaboliittiryhmään kuuluvaa lääkettä, joka estää tyrosiinin katabolian toista vaihetta: para-hydroksifenyylipyruvaatin muuttumista homogentisiinihapoksi. Tämän farmakologisen aineen käyttö johtaa kuitenkin tyrosiinin kertymiseen ja voi aiheuttaa vakavia sivuvaikutuksia, kuten sarveiskalvon samentumista ja valonarkuutta, nenäverenvuotoa ja mahaverenvuotoa, maksan vajaatoimintaa, muutoksia veressä jne. Yhdysvalloissa Nitisinoni on kuitenkin FDA:n hyväksymä tyypin I tyrosinemian hoitoon. [17 ], [ 18 ]
Alkaptonurian aiheuttamiin nivelongelmiin annetaan siis fysioterapiaa – liikuntaterapiaa lihasvoiman lisäämiseksi ja nivelten liikkuvuuden parantamiseksi, balneoterapiaa ja peloiditerapiaa kivun lievittämiseksi.
Vaikka tyrosiinia ei saada ainoastaan ruoasta, vaan sitä myös tuotetaan elimistössä, alkaptonuriaa sairastavien potilaiden suositellaan noudattavan vähäproteiinista ruokavaliota ja rajoittavan tyrosiinipitoisten ruokien, pääasiassa naudan- ja sianlihan, maitotuotteiden (erityisesti juustojen), palkokasvien, pähkinöiden ja siementen, kulutusta.
Ennaltaehkäisy
Geenimutaatioiden ehkäisy on mahdotonta, mutta synnynnäisten sairauksien riskin omaavien lasten syntymän estämiseksi on olemassa lääketieteellinen geneettinen neuvonta, joka on välttämätöntä ennen suunniteltua raskautta niille pariskunnille, joiden suvussa on perinnöllisiä sairauksia. [ 19 ]
Ennuste
Alkaptonurian aiheuttamat kuolemaan johtavat tulokset ovat hyvin harvinaisia, ja kuoleman voivat aiheuttaa vakavat sydän- ja munuaiskomplikaatiot. Alkaptonuriaa sairastavien ihmisten yleinen elinajanodote on siis hyvä.
Mutta elämänlaatu heikkenee nivelten tai selkärangan voimakkaan kivun vuoksi, johon liittyy merkittävä liikkuvuuden rajoittuminen, usein etenevästi.