Lääketieteen asiantuntija

Uudet julkaisut
Usherin oireyhtymä
Viimeksi tarkistettu: 04.07.2025

Kaikki iLive-sisältö tarkistetaan lääketieteellisesti tai se tarkistetaan tosiasiallisen tarkkuuden varmistamiseksi.
Meillä on tiukat hankintaohjeet ja vain linkki hyvämaineisiin mediasivustoihin, akateemisiin tutkimuslaitoksiin ja mahdollisuuksien mukaan lääketieteellisesti vertaisarvioituihin tutkimuksiin. Huomaa, että suluissa ([1], [2] jne.) Olevat numerot ovat napsautettavia linkkejä näihin tutkimuksiin.
Jos sinusta tuntuu, että jokin sisältö on virheellinen, vanhentunut tai muuten kyseenalainen, valitse se ja paina Ctrl + Enter.
Usherin oireyhtymä on perinnöllinen sairaus, joka ilmenee täydellisenä kuuroutena syntymästä lähtien sekä etenevänä sokeutumisena iän myötä. Näön heikkeneminen liittyy retinitis pigmentosa -tautiin, verkkokalvon pigmenttirappeumaan. Monilla Usherin oireyhtymää sairastavilla on myös vakavia tasapaino-ongelmia.
Epidemiologia
Tutkimuksen ansiosta voitiin todeta, että Usherin oireyhtymä vaikuttaa noin 8 prosenttiin tutkituista kuuromykistä lapsista (testit tehtiin kuuromykille tarkoitetuissa erityislaitoksissa). Pigmenttiretiniittiä havaittiin 6–10 prosentilla synnynnäisestä kuuroudesta kärsivistä potilaista, jota puolestaan havaitaan noin 30 prosentilla pigmenttiretiniittisairautta sairastavista.
Uskotaan, että tätä sairautta esiintyy maailmanlaajuisesti noin 3–10 ihmisellä 100 000:sta. Sitä esiintyy yhtä lailla sekä naisilla että miehillä. Noin 5–6 % maailman väestöstä kärsii tästä oireyhtymästä. Noin 10 % kaikista lapsuuden vaikeasta kuuroudesta johtuu Usherin oireyhtymästä I sekä tyypeistä II.
Yhdysvalloissa tyypit 1 ja 2 ovat yleisimmät. Yhdessä ne muodostavat noin 90–95 prosenttia kaikista Usherin oireyhtymän tapauksista lapsilla.
Syyt Usherin oireyhtymä
Usherin oireyhtymän tyypit I, II ja III periytyvät autosomissa peittyvästi, kun taas tyyppiä IV pidetään X-kromosomipoikkeavuutena. Tämän oireyhtymän yhteydessä esiintyvän sokeuden ja kuurouden syitä ei ole vielä tutkittu riittävästi. Oletetaan, että tätä sairautta sairastavat ihmiset ovat yliherkkiä DNA-rakennetta vaurioittaville komponenteille. Lisäksi tämä sairaus voi liittyä immuunijärjestelmän häiriöihin, mutta tässä tapauksessa tästä prosessista ei ole tarkkaa kuvaa.
Vuonna 1989 kromosomipoikkeavuuksia tunnistettiin ensimmäisen kerran tyypin II tautia sairastavilla potilailla, mikä voi tulevaisuudessa johtaa tapaan eristää oireyhtymää aiheuttavat geenit. Näitä geenejä voidaan myös mahdollisesti tunnistaa kantajilla ja kehittää erityisiä synnytystä edeltäviä geneettisiä testejä.
 [ 8 ]
[ 8 ]
Riskitekijät
Oireyhtymä periytyy, kun molemmat vanhemmat ovat sairaita, eli se periytyy resessiivisesti. Lapsi voi periä taudin myös, jos hänen vanhempansa ovat geenin kantajia. Jos molemmilla tulevilla vanhemmilla on tämä geeni, todennäköisyys saada vauva, jolla on tämä oireyhtymä, on 1/4. Henkilöä, jolla on vain yksi oireyhtymän geeni, pidetään kantajana, mutta hänellä ei ole sairauden oireita. Nykyään ei ole vielä mahdollista määrittää, onko henkilöllä tämän sairauden geeni.
Jos lapsi syntyy vanhemmille, joista toisella ei ole tällaista geeniä, oireyhtymän periytymisen todennäköisyys on hyvin pieni, mutta hän on varmasti kantaja.
Oireet Usherin oireyhtymä
Usherin oireyhtymän oireita ovat kuulon heikkeneminen ja pigmenttisolujen epänormaali kertyminen silmän rakenteisiin. Potilaalle kehittyy verkkokalvon rappeutuminen, joka aiheuttaa näön heikkenemistä ja vakavimmissa tapauksissa lopulta näön menetyksen.
Sensorineuraalinen kuulonalenema voi olla lievä tai täydellinen, eikä se yleensä etene syntymästä. Verkkokalvon pigmenttisairaus voi kuitenkin alkaa kehittyä lapsuudessa tai myöhemmin. Testitulokset ovat osoittaneet, että keskeinen näöntarkkuus voi säilyä useita vuosia, vaikka ääreisnäkö heikkenisi (tila, jota kutsutaan "tunnelinäköksi").
Nämä ovat taudin pääasialliset ilmenemismuodot, joita voivat joskus täydentää muut häiriöt, kuten psykoosi ja muut mielenterveyshäiriöt, sisäkorvan ongelmat ja/tai kaihi.
Lomakkeet
Tutkimuksen aikana tunnistettiin kolme taudin tyyppiä sekä neljäs muoto, joka on melko harvinainen.
Tyypin I sairaudelle on ominaista synnynnäinen täydellinen kuurous sekä tasapainohäiriö. Usein tällaiset lapset alkavat kävellä vasta 1,5 vuoden iässä. Näön heikkeneminen alkaa yleensä 10 vuoden iässä, ja hämäräsokeuden lopullinen kehitys alkaa 20 vuoden iässä. Tämän tyyppistä sairautta sairastavilla lapsilla voi kehittyä asteittainen perifeerisen näön heikkeneminen.
Tyypin II sairaudessa havaitaan kohtalaista tai synnynnäistä kuuroutta. Tässä tapauksessa osittaisen kuurouden heikkenemistä ei usein enää tapahdu. Pigmenttiretiniitti alkaa kehittyä murrosiän lopulla tai 20 vuoden iässä. Yösokeuden kehittyminen alkaa yleensä 29–31 vuoden iässä. Näöntarkkuuden heikkeneminen tyypin II patologiassa etenee yleensä hieman hitaammin kuin tyypin I sairaudessa.
Tyypin III taudille on ominaista etenevä kuulon heikkeneminen, joka yleensä alkaa murrosiässä, sekä samana aikana (hieman myöhemmin kuin kuulon heikkeneminen) asteittain kehittyvä retinitis pigmentosa, joka voi olla tekijä etenevän sokeuden kehittymisessä.
Tyypin IV patologian ilmenemismuodot esiintyvät pääasiassa miehillä. Tässä tapauksessa havaitaan myös eteneviä häiriöitä sekä kuulon ja näön heikkenemistä. Tämä muoto on hyvin harvinainen ja yleensä X-kromosomaalinen.
Diagnostiikka Usherin oireyhtymä
Usherin oireyhtymän diagnoosi tehdään potilaan havaitun äkillisen kuurouden ja etenevän näönmenetyksen yhdistelmän perusteella.
Testit
Mutaation havaitsemiseksi voidaan määrätä erityinen geneettinen testi.
Yksitoista geneettistä lokusta on löydetty, jotka voivat aiheuttaa Usherin oireyhtymän kehittymisen, ja yhdeksän geeniä on tunnistettu, jotka ovat ehdottomasti häiriön syy:
- Tyyppi 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Tyyppi 2: ush2a, VLGR1, WHRN.
- Usherin oireyhtymä tyyppi 3: USH3A.
NIDCD:n tutkijat ovat yhdessä New Yorkin ja Israelin yliopistojen kollegoidensa kanssa tunnistaneet Pcdh15-geenissä R245X-mutaation, joka selittää suuren osan tyypin 1 Usherin oireyhtymästä juutalaisväestössä.
Saadaksesi tietoa kliinisiä tutkimuksia suorittavista laboratorioista, käy osoitteessa https://www.genetests.org ja hae laboratoriohakemistosta "Usherin oireyhtymä".
Lisätietoja olemassa olevista kliinisistä tutkimuksista, joihin sisältyy Usherin oireyhtymän geenitestaus, saat osoitteesta https://www.clinicaltrials.gov hakemalla "Usherin oireyhtymä" tai "Usherin oireyhtymän geneettinen testaus".
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Instrumentaalinen diagnostiikka
Instrumentaaliseen diagnostiikkaan on useita menetelmiä:
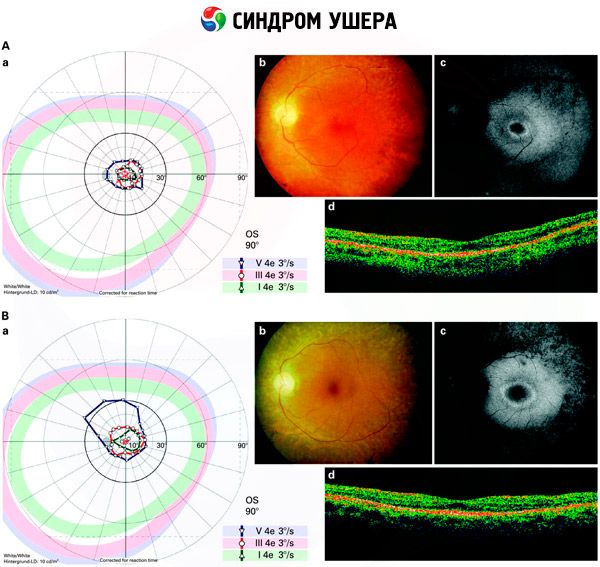
- Silmänpohjan tutkiminen verkkokalvon pigmenttipisteiden havaitsemiseksi sekä verkkokalvon verisuonten kaventuminen;
- Elektroretinogrammi, jonka avulla voidaan havaita silmän verkkokalvon alkuvaiheen degeneratiiviset poikkeamat. Se näyttää elektroradiografisten reittien sammumisen;
- Elektronistagmogrammi (ENG) mittaa tahattomia silmänliikkeitä, jotka voivat viitata epätasapainon olemassaoloon.
- Audiometria, jota käytetään kuurouden esiintymisen ja sen vakavuuden määrittämiseen.
Differentiaalinen diagnoosi
Usherin oireyhtymä on erotettava muista samankaltaisista sairauksista.
Hallgrenin oireyhtymä, jolle on ominaista synnynnäinen kuulon heikkeneminen ja etenevä näön heikkeneminen (myös kaihia ja nystagmusta kehittyy). Muita oireita ovat ataksia, psykomotoriset häiriöt, psykoosi ja kehitysvammaisuus.
Alströmin oireyhtymä, joka on perinnöllinen sairaus, jossa verkkokalvo rappeutuu ja johtaa keskeisen näön menetykseen. Tämä oireyhtymä liittyy lapsuusiän lihavuuteen. Samanaikaisesti diabetes mellitus ja kuulon heikkeneminen alkavat kehittyä 10 vuoden iän jälkeen.
Vihurirokko raskaana olevalla naisella ensimmäisen raskauskolmanneksen aikana voi aiheuttaa erilaisia poikkeavuuksia lapsen kehityksessä. Tällaisen poikkeavuuden seurauksiin kuuluvat kuulon heikkeneminen sekä (tai) näköongelmat ja tämän lisäksi erilaiset kehityshäiriöt.
Kuka ottaa yhteyttä?
Hoito Usherin oireyhtymä
Usherin oireyhtymään ei tällä hetkellä ole parannuskeinoa. Siksi hoito tässä tapauksessa koostuu pääasiassa näön heikkenemisen hidastamisesta sekä kuulon heikkenemisen kompensoimisesta. Mahdollisia hoitomenetelmiä ovat:
- A-vitamiinin ottaminen (jotkut silmälääkärit uskovat, että suuret A-vitamiinipalmitaattiannokset voivat hidastaa, mutta eivät pysäyttää, retinitis pigmentosan etenemistä);
- Erityisten elektronisten laitteiden istuttaminen potilaan korviin (kuulokojeet, sisäkorvaimplantit.
Silmälääkärit suosittelevat, että useimmat aikuiset, joilla on yleisiä retinitis pigmentosan muotoja, ottavat 15 000 IU (kansainvälistä yksikköä) A-vitamiinipalmitaattia päivittäin valvonnan alaisena. Koska tutkimukseen ei osallistunut tyypin 1 Usherin oireyhtymää sairastavia henkilöitä, suuria A-vitamiiniannoksia ei suositella tälle potilasryhmälle. A-vitamiinin käyttöä harkitsevien tulisi keskustella tästä hoitovaihtoehdosta lääkärinsä kanssa. Muita suosituksia tälle hoitovaihtoehdolle ovat:
- Ruokavalion muuttaminen siten, että se sisältää runsaasti A-vitamiinia sisältäviä ruokia.
- Raskautta suunnittelevien naisten tulisi lopettaa suurten A-vitamiiniannosten käyttö kolme kuukautta ennen suunniteltua raskautta lisääntyneen synnynnäisten epämuodostumien riskin vuoksi.
- Raskaana olevien naisten tulisi lopettaa suurten A-vitamiiniannosten käyttö lisääntyneen synnynnäisten epämuodostumien riskin vuoksi.
On myös tärkeää sopeuttaa tällainen lapsi sosiaaliseen elämään. Tämä vaatii erityisopettajien ja psykologien apua. Jos potilas on alkanut kokea asteittaista näköhäiriötä, hänelle tulisi opettaa viittomakielen käyttöä.
Ennuste
Usherin oireyhtymällä on epäsuotuisa ennuste. Näkökenttä ja sen terävyys alkavat heikentyä useimmilla potilailla 20–30 vuoden kuluessa. Joissakin tapauksissa esiintyy täydellinen molemminpuolinen näönmenetys. Kuulon heikkeneminen, johon aina liittyy mykkyys, kehittyy hyvin nopeasti täydelliseksi molemminpuoliseksi kuulon heikkenemiseksi.