Lääketieteen asiantuntija

Uudet julkaisut
Angelmanin oireyhtymä lapsilla ja aikuisilla
Viimeksi tarkistettu: 04.07.2025

Kaikki iLive-sisältö tarkistetaan lääketieteellisesti tai se tarkistetaan tosiasiallisen tarkkuuden varmistamiseksi.
Meillä on tiukat hankintaohjeet ja vain linkki hyvämaineisiin mediasivustoihin, akateemisiin tutkimuslaitoksiin ja mahdollisuuksien mukaan lääketieteellisesti vertaisarvioituihin tutkimuksiin. Huomaa, että suluissa ([1], [2] jne.) Olevat numerot ovat napsautettavia linkkejä näihin tutkimuksiin.
Jos sinusta tuntuu, että jokin sisältö on virheellinen, vanhentunut tai muuten kyseenalainen, valitse se ja paina Ctrl + Enter.

On olemassa useita sairauksia, joiden kohdalla ilmaisut kuten "pidä huolta itsestäsi, niin et sairastu" kuulostavat vähintäänkin naurettavilta. Nämä ovat patologioita, joissa lapsen kehossa on jo ennen syntymää joitakin henkisiä ja fyysisiä poikkeavuuksia, mutta vanhemmat eivät ole tästä syypäitä. Tällaiset sairaudet johtuvat kromosomistojen mutaatioista tai poikkeavuuksista, ja niitä kutsutaan kromosomisairauksiksi tai geneettisiksi. Angelmanin oireyhtymä, Downin oireyhtymä, Pataun oireyhtymä, Edwardsin oireyhtymä, Turnerin oireyhtymä, Prader-Willin oireyhtymä - tämä on vain osa geneettisistä sairauksista melko kohtuullisen listan joukosta.
Onnellisen miehen oireyhtymä
Tällä kertaa puhumme englantilaisen lastenlääkärin Harry Angelmanin mukaan nimetystä patologiasta. Angelman nosti ongelman ensimmäisen kerran esiin vuonna 1965 kohdattuaan vastaanotollaan edellisenä päivänä kolme epätavallista lasta, joita yhdistivät yhteiset erikoiset oireet. Lääkäri kutsui näitä lapsia nukkelapsiksi ja kirjoitti heistä artikkelin, jonka alkuperäinen nimi oli "Lapset-marionetit". Itse artikkeli ja sen otsikko kirjoitettiin Veronan museoissa nähdyn maalauksen innoittamana. Maalaus kuvasi nauravaa poikaa, ja sitä kutsuttiin nimellä "Nukkepoika". Maalauksessa kuvatun lapsen yhteys kolmeen lapseen, jotka Angelman oli kerran tavannut vastaanotollaan, sai lastenlääkärin yhdistämään lapset yhdeksi ryhmäksi heidän sairautensa vuoksi.
Ei ole mitään yllättävää siinä, etteivät muut lääkärit huomanneet artikkelissa mainittuja lapsia. Ensi silmäyksellä näytti siltä, että heillä oli täysin erilaiset sairaudet, joten taudin yleinen kliininen kuva kolmessa eri tapauksessa oli niin erilainen. Ehkä "uusi" kromosomipatologia olisi kiinnostanut muita tiedemiehiä, mutta tuolloin genetiikka ei ollut vielä tarpeeksi kehittynyt vahvistamaan englantilaisen lääkärin hypoteesia. Siksi artikkeli heitettiin takahyllylle pitkäksi aikaa, kun sitä kohtaan tunnettiin tiettyä kiinnostusta.
Seuraava maininta Angelmanin oireyhtymästä, jota englantilaisen lastenlääkärin G. Angelmanin artikkeli nyt kutsuttiin nimellä, juontaa juurensa 1980-luvun alkuun. Ja vasta vuonna 1987 löydettiin syy siihen, miksi pieni osa lapsista syntyy sellaisilla poikkeamilla, että ulkopuolelta he näyttävät jatkuvasti hymyileviltä ja onnellisilta. Itse asiassa tämä ei pidä paikkaansa ollenkaan, ja hymy on vain irvistys, jonka takana piiloutuu onneton ihmissielu ja vanhempien tuska.
Epidemiologia
Tilastojen mukaan lapsen kromosomimutaatio voi kehittyä sekä vanhempien samankaltaisten mutaatioiden taustalla että ilman niitä. Angelmanin oireyhtymällä (AS) ei ole selkeää perinnöllistä luonnetta, mutta patologian kehittymisen todennäköisyys vanhemmilla, joilla on kromosomimutaatioita, on melko korkea.
On myös mielenkiintoista, että jos perheessä on jo AS-lapsi, on yhden prosentin mahdollisuus saada toinen lapsi, jolla on sama häiriö, vaikka vanhemmat olisivatkin terveitä.
Angelmanin oireyhtymää sairastavien potilaiden määrästä ei ole vielä tarkkoja tilastoja. Ehkä syynä on oireiden monimuotoisuus, jotka voivat esiintyä tietyssä koossa tai olla esiintymättä lainkaan pitkään aikaan. Oletetaan, että taudin esiintyvyys on: 1 lapsi 20 000 vastasyntynyttä kohden. Mutta tämä luku on hyvin likimääräinen.
Syyt Angelmanin oireyhtymä
Angelmanin oireyhtymä on lääketieteellinen nimi kromosomipoikkeavuudelle, mutta se on kaikkea muuta kuin ainoa. Ihmiset kutsuvat tätä sairautta nukkelapsisyndroomaksi, onnellisen nukkesyndroomaksi, Petruškan oireyhtymäksi ja nauravan nuken oireyhtymäksi. Ihmiset keksivät kaikenlaisia nimiä (joskus jopa loukkaavia potilaille itselleen ja heidän vanhemmilleen), mutta sairaus on sairaus, olipa se kuinka hauskalta tahansa ja syyt mitkä tahansa.
Ja Angelmanin oireyhtymän kehittymisen syyt, kuten monet muutkin geneettiset sairaudet, ovat kaikissa tapauksissa yhden kromosomin tai koko kromosomijoukon rakenteen häiriöt. Mutta meidän tapauksessamme koko ongelma on äidiltä periytyvässä kromosomissa 15. Eli isän kromosomissa ei tässä tapauksessa ole poikkeamia, mutta naisen kromosomissa tapahtuu tiettyjä mutaatioita.
Kromosomipoikkeavuuden tyypin mukaan Angelmanin oireyhtymä luokitellaan kromosomimutaatioksi. Tällaisia mutaatioita pidetään:
- Deleetio (tietyn geenijoukon sisältävän kromosomin osan puuttuminen; jos yksi geeneistä puuttuu, puhutaan mikrodeleetiosta), joka on seurausta kahdesta katkeamisesta ja yhdestä yhdistymisestä, kun osa alkuperäisestä kromosomista menetetään.
- Kopiointi (ylimääräisen osan läsnäolo kromosomissa, joka on kopio olemassa olevasta), joka useimmissa tapauksissa johtaa henkilön kuolemaan ja harvemmin hedelmättömyyteen.
- Inversio (kromosomin yhden osan kääntyminen 180 astetta eli vastakkaiseen suuntaan, jolloin siinä olevat geenit sijaitsevat vastakkaisessa järjestyksessä), kun kromosomin katkenneet päät liittyvät toisiinsa eri järjestyksessä kuin alkuperäinen.
- Lisäys (jos osa kromosomin geneettisestä materiaalista on pois paikaltaan),
- translokaatio (jos tietty kromosomin osa on kiinnittynyt toiseen kromosomiin; tällainen mutaatio voi olla molemminpuolinen ilman osioiden menetystä).
Saatuaan mutatoituneen kromosomin tietämättömältä äidiltä lapsi on tuomittu syntymään poikkeavuuksin. Angelmanin oireyhtymän yleisimpänä syynä pidetään edelleen äidin 15. kromosomin deleetiota, jossa pieni osa puuttuu. Harvinaisempia mutaatioita "nauravan nuken" oireyhtymässä pidetään:
- translokaatio
- unipaternaalinen disomia (jos lapsi sai isältään kromosomiparin, äidin kromosomi puuttuu),
- DNA:n geenien mutaatio, jotka ovat sekä tärkein rakennusmateriaali (geneettinen materiaali) että ohjeet sen oikealle käytölle (erityisesti ube3a-geenin mutaatio äidin kromosomissa).
Jonkin näistä mutaatioista esiintyminen vanhemmilla on riskitekijä Angelmanin oireyhtymän kehittymiselle lapsilla. Mutta kromosomimutaatioiden lisäksi myös genomiset mutaatiot (jotka liittyvät kromosomistojen määrälliseen muutokseen ja ovat yleisempiä kuin kromosomimutaatiot) voivat laukaista taudin kehittymisen lapsella. Yleisiä genomisia mutaatioita ovat kromosomitrisomia (jos henkilön kromosomistossa on yli 46 kromosomia).
Jotta patologia ilmenisi lapsella, ei ole lainkaan välttämätöntä, että vanhemmilla on kromosomipoikkeavuuksia. Silti on tietty prosenttiosuus potilaista, joilla sairaus on perinnöllinen.
Synnyssä
Sukellataanpa hieman syvemmälle biologiaan eli tarkemmin sanottuna genetiikkaan. Jokaisen ihmisorganismin geneettinen informaatio sisältyy 23 kromosomipariin. Parista toinen kromosomi periytyy lapselle isältä, toinen äidiltä. Kaikki kromosomiparit eroavat toisistaan muodoltaan ja kooltaan ja sisältävät tiettyä tietoa. Siten 23. kromosomipari (X- ja Y-kromosomit) vastaa vauvan seksuaalisten ominaisuuksien muodostumisesta (XX - tyttö, XY - poika, kun taas Y-kromosomin lapsi voi saada vain isältä).
Ihannetapauksessa lapsi saa vanhemmiltaan 46 kromosomia, jotka muodostavat hänen geneettiset ominaisuutensa ja määräävät hänet yksilönä. Suurempaa kromosomimäärää kutsutaan trisomiaksi ja sitä pidetään poikkeamana normista. Esimerkiksi kromosomin 47 esiintyminen kromosomistossa (karyotyyppi, joka määrittää lajin ja yksilölliset ominaisuudet) aiheuttaa Downin syndrooman esiintymisen.
Jos kromosomit värjätään erityisellä väriaineella, mikroskoopilla voi nähdä eri sävyisiä raitoja kutakin niistä pitkin. Kunkin raidan sisällä on valtava määrä geenejä. Kaikki nämä raidat ovat tiedemiesten numeroimia ja niillä on kiinteä sijainti. Yhden raidan puuttumista pidetään poikkeamana normista. Angelmanin oireyhtymässä voidaan hyvin usein havaita äidin kromosomin segmenttien puuttuminen q11-q13-välillä, joka sijaitsee pitkässä haarassa, jossa DNA-emästen määrä on vain noin 4 miljoonaa.
Kromosomin pääkomponenttina pidetään uskomattoman pitkää DNA-molekyyliä, joka sisältää tuhansia geenejä ja kymmeniä ja satoja miljoonia typpipitoisia emäksiä. Niinpä kromosomi 15, joka on vastuussa Angelmanin oireyhtymän ja useiden muiden sairauksien kehittymisestä, sisältää 1200 geeniä ja noin 100 miljoonaa emästä. Kaikki DNA-molekyylin rakenteen häiriöt vaikuttavat varmasti tulevan lapsen ulkonäköön ja kehitykseen.
Geenien sisältämä geneettinen informaatio muunnetaan proteiiniksi eli RNA:ksi. Tätä prosessia kutsutaan geenien ilmentymiseksi. Tällä tavoin vanhemmilta saatu geneettinen informaatio saa sekä muodon että sisällön, joka ilmentyy heidän ainutlaatuisessa nais- tai miesperillisessään.
On olemassa useita patologioita, joilla on ei-klassinen perintötyyppi, mukaan lukien Angelmanin oireyhtymä, jossa vanhemmilta paritettujen kromosomien osana saadut geenit kantavat ainutlaatuista vanhempien jälkiä ja ilmenevät eri tavoin.
Angelmanin oireyhtymä on siis silmiinpistävä esimerkki genomisesta painautumisesta, jossa geenien ilmentyminen lapsen kehossa riippuu suoraan siitä, kummalta vanhemmalta alleelit on saatu (saman geenin eri muodot, jotka on saatu isältä ja äidiltä, sijaitsevat identtisissä parillisten kromosomien osissa). Toisin sanoen vain äidin kromosomin poikkeavuudet johtavat oireyhtymän kehittymiseen, kun taas isän kromosomin mutaatiot ja rakenteelliset häiriöt aiheuttavat täysin erilaisia patologioita.

Tässä patologiassa äidin kromosomissa puuttuu tiettyjä geenejä tai yksittäisten geenien aktiivisuus on vähentynyt/katoaa (useimmissa tapauksissa ube3a-geeni, joka osallistuu ubikitiini-nimisen proteiinin metaboliaan, joka säätelee muiden proteiinien hajoamista). Tämän seurauksena lapsella diagnosoidaan henkisen kehityksen poikkeavuuksia ja fyysisiä epämuodostumia.
Oireet Angelmanin oireyhtymä
Angelmanin oireyhtymän oireet vaikuttavat lapsen elämän ja kehityksen eri osa-alueisiin: fyysiseen, neurologiseen ja henkiseen. Tämän perusteella voidaan tunnistaa kolme oireryhmää, jotka viittaavat tämän patologian kehittymiseen.
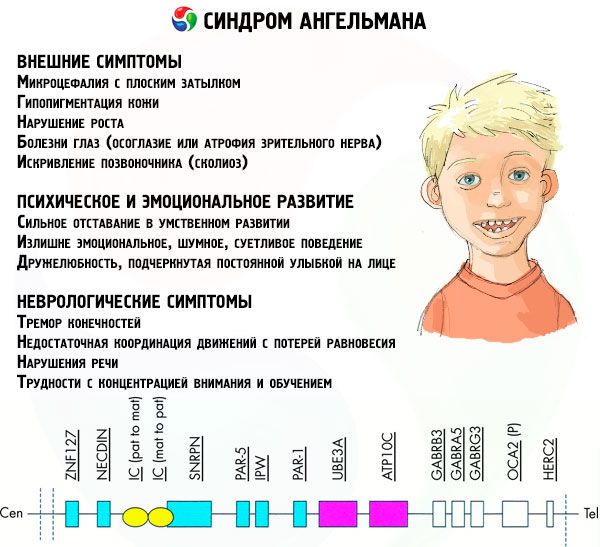
- Ulkoiset tai fyysiset oireet:
- suhteettoman pieni pää verrattuna normaalin kokoiseen vartaloon ja raajoihin,
- liian leveä suu,
- kasvoilla on melkein aina hymy (suu auki),
- harvat hampaat,
- kapea ylähuuli,
- usein ulkoneva leveä kieli,
- ulkoneva alaleuka,
- terävä leuka,
- hyvin vaalea iho, usein karvat (albinismi, joka liittyy siihen, että keho ei tuota melaniinipigmenttiä),
- tummat läiskät vaalealla iholla (hypopigmentaatio riittämättömän melaniinin tuotannon vuoksi)
- fyysisiä tai ulkoisia oireita: silmäsairauksia, kuten strabismus tai näköhermon surkastumista,
- selkärangan kaarevuus (skolioosi),
- jäykät jalat (kävellessään henkilö ei taivuta jalkojaan polvissa nivelten alhaisen liikkuvuuden vuoksi, joten vertailu nuken kävelyyn).
- Henkiseen ja emotionaaliseen kehitykseen liittyvät oireet:
- vaikea kehitysvammaisuus,
- ylitunteellinen, meluisa ja kiukkuinen käytös,
- usein käsien taputtelua,
- ilmaistu ystävällisyys, jota korostaa jatkuva hymy kasvoilla,
- usein naurua ilman syytä.
- Neurologiset oireet:
- raajojen vapina,
- liikkeiden riittämätön koordinointi tasapainon menetykseen,
- vähentynyt lihasjänteys,
- erilaisia unihäiriöitä,
- usein hysteerisiä kohtauksia lapsuudessa,
- puhehäiriöt (lapsi alkaa puhua myöhään, hänellä on heikot kommunikointitaidot ja epäselvä puhe),
- hyperaktiivisuus lisääntyneen herkkyyden taustalla,
- keskittymis- ja oppimisvaikeuksia.
Mutta tämä on yleistetty kuva taudista. Itse asiassa Angelmanin oireyhtymän kliininen kuva riippuu pitkälti taudin kehitysvaiheesta ja patologian aiheuttaneen kromosomimutaation tyypistä. Tämä tarkoittaa, että taudin oireet voivat vaihdella merkittävästi eri potilailla, mikä pitkään ei antanut meille mahdollisuutta erottaa patologiaa muista, joilla on samanlainen kliininen kuva.
Kokonaismäärästä oireita voidaan erottaa ne, jotka ovat poikkeuksetta tyypillisiä kaikille potilaille:
- vaikea kehitysvammaisuus,
- sopimaton käyttäytyminen (kohtuuton nauru, lisääntynyt kiihtyvyys, huono keskittymiskyky, euforian tila),
- motoristen taitojen alikehittyminen,
- huono liikkeiden koordinaatio, kävelyataksia (epätasainen tahti, huojuminen puolelta toiselle jne.), raajojen vapina.
- puheenkehityshäiriö, jossa vallitsee ei-verbaalinen viestintä.
Suurimmalla osalla potilaista esiintyvistä oireista voidaan erottaa seuraavat:
- pään ja vartalon välinen epäsuhta, joka johtuu viivästyneestä fyysisestä kehityksestä,
- monilla potilailla kallon muoto on sellainen, että aivojen koko pysyy pienempänä kuin terveillä ihmisillä (mikrokefalia),
- epileptiset kohtaukset ennen 3 vuoden ikää, joiden voimakkuus ja esiintymistiheys vähenevät asteittain vanhemmalla iällä,
- EEG-parametrien vääristymä (matalataajuisten aaltojen vaihtelut ja suuri amplitudi).
Nämä oireet ovat melko yleisiä, mutta 20 prosentilla Angelmanin oireyhtymää sairastavista potilaista niitä ei ole.
Vielä harvemmin on mahdollista diagnosoida taudin tällaisia ilmenemismuotoja:
- vaikea tai lievä strabismus,
- huono kielen liikkeen hallinta, minkä seurauksena potilaat usein työntävät kieltään ulos ilman syytä,
- nielemis- ja imemisvaikeudet, erityisesti pienillä lapsilla,
- ihon ja silmien pigmentaation häiriintyminen,
- kävellessä kävelemällä kävelemällä ylös tai koukussa
- hyperrefleksia,
- unihäiriöt, erityisesti lapsuudessa,
- tiheä syljeneritys,
- kyltymätön jano,
- liian aktiiviset pureskeluliikkeet,
- yliherkkyys kuumuudelle,
- litteä pään takaosa,
- ulkoneva alaleuka,
- sileät kämmenet.
Melko suurella osalla potilaista on virtsaamisvaikeuksia, joita he hallitsevat huonosti, heikentyneitä hienomotorisia taitoja, jotka vaikeuttavat itsehoitoa ja oppimista, sekä ylipainoa. Lähes kaikki potilaat kokevat murrosiän myöhemmin kuin terveet ikätoverit.
Angelmanin oireyhtymää sairastavat lapset havaitsevat suullisen puheen hyvin ja ymmärtävät sitä, mutta eivät halua osallistua keskusteluun, rajoittaen puheensa muutamaan kymmeneen sanaan, jotka ovat välttämättömiä jokapäiväisessä elämässä. Aikuisuudessa tällaiset potilaat näyttävät kuitenkin nuoremmilta kuin ikätoverinsa, joilla ei ole geneettisiä patologioita.
Monet Angelmanin oireyhtymän oireet ovat epäsäännöllisiä, joten taudin kliininen kuva muuttuu merkittävästi iän myötä. Kouristukset ja epileptiset kohtaukset harvenevat tai häviävät kokonaan, potilas vähenee ja uni paranee.
Komplikaatiot ja seuraukset
Angelmanin oireyhtymä on vakava, tällä hetkellä käytännössä parantumaton kromosomipoikkeavuus, joka estää potilaita elämään normaalia elämää. AS-oireyhtymää sairastavan lapsen elämä riippuu pitkälti kromosomipoikkeavuuden tyypistä.
Kromosomisegmentin kahdentuminen on useimmissa tapauksissa yhteensopimaton elämän kanssa. Ja vaikka tällaiset potilaat eivät kuolisikaan imeväisikäisenä ja saavuttaisi murrosikää, heillä ei ole mahdollisuutta saada lapsia.
Angelmanin oireyhtymässä useimmin esiintyvien geenien osan deleetio tai puuttuminen on este lapsen kävelyn ja puhumisen oppimiselle. Tällaisilla lapsilla on vaikeampi kehitysvammaisuus, ja epileptisiä kohtauksia esiintyy useammin ja niiden voimakkuus on paljon suurempi kuin potilailla, joilla on muita kromosomipoikkeavuuksia.
Jos vain yhden geenin mutaatio on olemassa, lapselle voidaan asianmukaisella huomiolla ja lähestymistavalla opettaa itsestä huolehtimisen, kommunikoinnin ja ryhmässä vuorovaikutuksen perusteet, vaikka hän jää silti kehityksessä jälkeen ikätovereistaan.
Luonteeltaan ystävällisille Angelmanin oireyhtymää sairastaville lapsille tärkeintä on vanhempien rakkaus ja huomio. Vain tässä tapauksessa lapsen koulutus kantaa hedelmää, vaikka se olisikin vähäistä. Tietenkin AS-potilaat eivät pysty opiskelemaan tavallisessa koulussa. He tarvitsevat erityisluokkia, joissa lapsille opetetaan ensin keskittymistä ja sitten vähitellen annetaan koulutiedon perusteet.
Diagnostiikka Angelmanin oireyhtymä
Angelmanin oireyhtymä on synnynnäinen kehityshäiriö. Mutta tietyistä olosuhteista johtuen sitä on usein mahdotonta diagnosoida imeväis- ja varhaislapsuudessa. Tämä johtuu oireiden epäspesifisyydestä ja heikosta ilmentymisestä imeväisillä ja alle 3-vuotiailla lapsilla. Eikä taudin esiintyvyys maassamme ole niin suuri, että lääkärit olisivat oppineet tunnistamaan sen vertaistensa joukossa.
Angelmanin oireyhtymä voi ilmetä imeväisillä lihasjänteyden heikkenemisenä, mikä ilmenee syömisongelmina (imemis- ja nielemisrefleksin heikkous) ja myöhemmin vaikeuksina kävelemisen oppimisessa (tällaiset lapset alkavat kävellä paljon myöhemmin). Nämä oireet ovat ensimmäisiä merkkejä vauvan kehityshäiriöstä, joka voi hyvinkin liittyä kromosomipoikkeavuuteen. Vain geneettinen analyysi voi vahvistaa tämän oletuksen.
Erityistä huomiota kiinnitetään lapsiin, joiden vanhemmilla on erilaisia genomisia tai kromosomipoikkeavuuksia. Tauti ei nimittäin välttämättä ilmene aluksi, ja jos se havaitaan ajoissa ja lapsen kanssa intensiivisen työskentelyn aloittaminen voi johtaa merkittävästi parempaan oppimismenestykseen ja taudin etenemisen hidastumiseen.
Jos vanhemmilla on erilaisia kromosomipoikkeavuuksia, geneettinen analyysi suoritetaan jo ennen vauvan syntymää, koska SA on yksi alkionvaiheessa havaittavista patologioista.
Geenitutkimuksen aineiston kerääminen voidaan suorittaa kahdella tavalla:
- invasiivinen (tietyllä riskiprosentilla, koska on välttämätöntä tunkeutua kohtuun lapsiveden näytteen ottamiseksi),
- ei-invasiivinen (vauvan DNA:n analyysi äidin verestä).
Sitten suoritetaan seuraavat tutkimukset:
- fluoresoiva in situ -hybridisaatio (FISH-menetelmä) – erityisellä väriaineella leimatun DNA-koettimen sitoutuminen tutkittavaan DNA:han, minkä jälkeen tutkimus mikroskoopilla.
- ube3a-geenin ja painettujen geenien mutaatioiden analyysi,
- DNA-metylaatioanalyysi genetiikassa käytettyjä erityismenetelmiä käyttäen.
Geenitestit antavat melko tarkkaa tietoa kromosomipoikkeavuuksien tapauksessa, mikä tarkoittaa, että tulevat vanhemmat tietävät etukäteen, mihin varautua. Poikkeuksia kuitenkin on. Tietyllä potilasryhmällä, vaikka kaikki patologiaan viittaavat oireet ilmenisivät, testitulokset pysyvät normaaleina. Eli patologia voidaan tunnistaa vain tarkkailemalla lasta huolellisesti varhaislapsuudesta lähtien: miten hän syö, milloin hän alkoi kävellä ja puhua, koukistaako hän jalkojaan kävellessään jne.
FISH-menetelmän lisäksi Angelmanin oireyhtymän instrumentaalisista diagnostisista menetelmistä voidaan erottaa tomografia (CT tai MRI), joka auttaa määrittämään aivojen tilan ja koon, sekä aivosähkökäyrä (EEG), joka osoittaa, miten aivojen yksittäiset osat toimivat.
Lääkärit tekevät lopullisen diagnoosin yleensä 3–7 vuoden iässä, kun potilaalla on jo suurin osa oireista ja taudin kehityksen dynamiikka on näkyvissä.
Mitä testejä tarvitaan?
Differentiaalinen diagnoosi
Angelmanin oireyhtymä on geneettinen patologia, jolla ei käytännössä ole erityisiä ilmenemismuotoja. Useimmat oireet voivat yhtä lailla viitata sekä AS:ään että muihin geneettisiin patologioihin.
Angelmanin oireyhtymän differentiaalinen diagnoosi suoritetaan seuraavilla patologioilla:
- Pitt-Hopkinsin oireyhtymä (potilaille on ominaista kehitysvammaisuus, iloinen luonne, hymyilevä, heillä on melko suuri ja leveä suu, havaitaan mikrokefaliaa). Ero on hyperventilaatiokohtaukset ja hengityksen pidättäminen valveilla ollessa.
- Christiansonin oireyhtymä (potilaat ovat henkisesti jälkeenjääneitä ihmisiä, joilla on iloinen luonne, jotka eivät kykene puhumaan, joille on ominaista mikrokefalia, ataksia, kouristukset, tahattomat lihasliikkeet).
- Mowat-Wilsonin oireyhtymä (oireita: kehitysvammaisuus, epileptiset kohtaukset, terävä leuka, avoin suu, onnellinen ilme kasvoilla, mikrokefalia). Tunnusmerkkejä: suuri silmien välinen etäisyys, sisäänpäin vinot silmät, pyöreä nenänpää, taaksepäin kiertynyt korvalehti.
- Kabuki-oireyhtymä (jolle on ominaista lievä tai kohtalainen kehitysvammaisuus, puhe- ja motoriset ongelmat, lihasheikkous, epileptiset kohtaukset, mikrokefalia, pitkät kutinavälit ja koordinaatiohäiriöt). Oireyhtymälle ovat kaarevat kulmakarvat, alaluomen ulospäin kääntynyt sivuosa, kaukana toisistaan sijaitsevat silmät, pitkät luomihalkeamat ja pitkät, paksut silmäripset.
- Rettin oireyhtymä (erottelu AS:stä naisilla). Oireet: viivästynyt puheenkehitys, kouristuskohtaukset, mikrokefalia. Ero on siinä, että kasvoilla ei ole iloista ilmettä, esiintyy uniapnea- ja apraksiakohtauksia, jotka etenevät ajan myötä.
- Autosomaalinen peittyvä kehitysvammaisuusoireyhtymä 38 (oireet: huomattava kehitysvammaisuus, johon liittyy viiveitä motorisissa taidoissa ja puheessa, lihasheikkous, syömisongelmia imeväisillä, impulsiivisuus). Tyypillinen piirre on iiriksen sininen väri.
- MECP 2 -geenin duplikaatio-oireyhtymä (erilaistuminen SA:sta miehillä). Oireet: vaikea kehitysvammaisuus, lihasheikkous lapsuudesta lähtien, puheongelmat tai puhekyvyn puute, epilepsia. Määritelmät: etenevä myopatia, jatkuvasti toistuvat infektiot.
- Kleefstran oireyhtymä (oireita: puhe- ja ajatteluhäiriöt, lihasheikkous, unihäiriöt, tarkkaavuuden puute, avoin suu, hyperaktiivisuus, kouristuskohtaukset, ataksia, tasapainohäiriöt). Tunnusmerkkejä: litteä kasvot, lyhyt tylppä nenä, etäällä sijaitsevat silmät, suuri, ulospäin kääntynyt alahuuli, aggressiiviset purkaukset.
- Smith-Magenisin oireyhtymä (jolle on ominaista kouristuskohtaukset, unihäiriöt, älyllisen ja motorisen kehityksen häiriöt). Tyypillisiä piirteitä ovat leveät ja litteät kasvot sekä erottuva otsa.
- Koolen-de-Vriesin oireyhtymä (lievä tai kohtalainen kehitysvammaisuus, lihasheikkous, kouristuskohtaukset, ystävällisyys). Tunnusmerkkejä: pitkänomainen kasvot korkealla otsalla, ulkonevat korvat, vinot silmät, suuri nivelten liikkuvuus, synnynnäiset sydänviat.
- Phelan-McDermidin oireyhtymä (oireet: kehitysvammaisuus, puhehäiriöt tai puhekyvyn puute). Oireet: suuret kädet kehittyneillä lihaksilla, syntymästä lähtien ilmennyt lihasheikkous, heikko hikoilu.
Tällaiset patologiat, kuten adenyylisukkinaatin puutos, autosomaalinen peittyvä kehitysvammaisuusoireyhtymä 1, kromosomi 2q23.1 -duplikaatio-oireyhtymä, FOXG1-, STXBP1- tai MEF2C-geenin haploinsuffisienssioireyhtymät ja jotkut muut, voivat "ylpeillä" Angelmanin oireyhtymän kaltaisilla oireilla.
Lääkärin tehtävänä on tehdä tarkka diagnoosi, erottaa Angelmanin oireyhtymä samankaltaisista oireista kärsivistä patologioista ja määrätä tehokas hoito, joka on merkityksellinen taudin diagnosoidulle vaiheelle.
Kuka ottaa yhteyttä?
Hoito Angelmanin oireyhtymä
Angelmanin oireyhtymä on yksi niistä patologioista, joihin lääketiede etsii edelleen tehokasta hoitoa. Taudin etiologinen hoito on kehitysvaiheessa useilla menetelmillä ja keinoilla, joista monia ei ole vielä testattu ihmisillä. Tämä tarkoittaa, että toistaiseksi lääkäreiden on rajoituttava oireenmukaiseen hoitoon, joka auttaa jollain tavalla lievittämään marionettioireyhtymästä kärsivien lasten ja aikuisten kadehdittavaa tilannetta, sillä he kärsivät epileptisistä kohtauksista, syljenerityksestä, hypotensiosta ja unihäiriöistä.
Näin ollen on mahdollista vähentää epileptisten kohtausten esiintymistiheyttä ja voimakkuutta oikein valitun kouristuslääkkeen avulla. Mutta koko vaikeus piilee siinä, että SA-potilaiden kohtaukset eroavat tavallisista epileptisistä kohtauksista siinä, että niille on ominaista useat kohtaustyypit, mikä tarkoittaa, että tilaa voidaan lievittää antamalla useita lääkkeitä kerralla.
Angelmanin oireyhtymän hoitoon käytetyistä kouristuslääkkeistä suosituimpia ovat valproiinihappo, topiramaatti, lamotrigiini, levetirasetaami, klonatsepaami ja niihin perustuvat lääkkeet. Harvemmin käytetään karmatsepiiniin, fenytoiiniin, fenobarbitaaliin ja etosuksimidiin perustuvia lääkkeitä, koska jotkut niistä voivat aiheuttaa paradoksaalisen vaikutuksen, joka koostuu epileptisten kohtausten voimistumisesta ja tihentymisestä. Tämä tapahtuu, jos lääkettä käytetään osana monoterapiaa.
Kuolaamisen hoitoon käytetään yleensä kahta menetelmää: lääkehoitoa (syljen tuotantoa vähentävät lääkkeet) ja kirurgista hoitoa, jossa sylkitiehyet asennetaan uudelleen. Mutta sylkirauhasten limakalvojen...
Toinen ongelma on lyhyt unen kesto. Usein Angelmanin oireyhtymää sairastavat lapset nukkuvat enintään 5 tuntia, mikä vaikuttaa negatiivisesti koko kehon toimintaan. Helposti innostuvat, aktiiviset lapset, jotka rakastavat pelejä ja kommunikointia (vaikka yrittäisivätkin rajoittua sanattomiin menetelmiin), ovat huomattavan väsyneitä päivän aikana. Jotta keho saisi hyvän levon, se tarvitsee syvää ja täyttä unta, mutta juuri tämä on ongelma.
Vaikuttaa siltä, että hermostoa rauhoittavat rauhoittavat lääkkeet (fenotiatsiinit ja epätyypilliset antipsykootit) riittävät parantamaan unta herkillä potilailla. Mutta AS:n tapauksessa tällaisten lääkkeiden käyttöön liittyy suuri negatiivisten vaikutusten riski. Siksi lääkärit suosivat edelleen lieviä unilääkkeitä, kuten melatoniinia (luonnollinen unihormoniin perustuva hormonaalinen lääke), jota annetaan potilaille tunti ennen nukkumaanmenoa yhden tabletin annoksena, ja difenhydramiinia. Lääkäri määrää lääkkeen antotiheyden ja annostuksen potilaan tilan ja iän perusteella.
Angelmanin oireyhtymää sairastavilla potilailla on joskus ongelmia ruoansulatuksen ja ulostamisen kanssa. Ulostetta voi parantaa laksatiiveilla (mieluiten rohdosvalmisteilla).
Tai voit lähestyä ongelmaa eri tavalla, kuten amerikkalaiset lääkärit tekivät, perustuen joihinkin autismin hoitomenetelmiin, koska monet AS:lle tyypilliset oireet ovat myös tyypillisiä autismille (impulsiivisuus, tahattomat liikkeet, toistuvat toiminnot, tarkkaavaisuusvaje, kommunikaatio-ongelmat jne.). Todettiin, että sekretiinin hormonin käyttöönotto, joka normalisoi ruoansulatusta ja ulostamista, vaikuttaa positiivisesti potilaiden tarkkaavaisuuteen, ja oksitosiini auttaa parantamaan lapsen kognitiivisia kykyjä ja muistia sekä korjaamaan käyttäytymistä.
Pelkät hormonit eivät tosin riitä, varsinkaan lasten kohdalla. Angelmanin oireyhtymässä käytetään käyttäytymisterapiaa, työskentelyä psykologin ja puheterapeutin kanssa (ei-verbaalisen viestinnän menetelmien ja viittomakielen opettaminen). Tällaisten lasten koulutuksen tulisi perustua yksilölliseen ohjelmaan, johon osallistuvat erityiskoulutetut opettajat, psykologi ja vanhemmat. Valitettavasti tämä ei ole mahdollista kaikkialla, ja perheet jäävät yksin ongelmansa kanssa.
Koska monet nuoret AS-potilaat kärsivät alhaisesta lihasjänteyksestä ja nivelongelmista, fysioterapiaan kiinnitetään paljon huomiota. Useimmiten lääkärit turvautuvat parafiinihoitoihin, elektroforeesiin ja magneettiterapiaan.
Aktiivinen tonic-hieronta ja erityiset terapeuttisen fyysisen harjoittelun harjoitukset auttavat sairasta lasta seisomaan jaloilleen ja kävelemään luottavaisesti jonkin ajan kuluttua. Tässä suhteessa erityisen hyödyllinen on vesivoimistelu, jota suositellaan kylmässä vedessä liikkuville. Se lisää lihastonusta ja opettaa lasta hallitsemaan kehoaan ja koordinoimaan liikkeitään.
Kouristuksia estävä hoito
Angelmanin oireyhtymän vaarallisin oire on epilepsian kaltaiset kohtaukset. Tätä oiretta esiintyy 80 %:lla potilaista, mikä tarkoittaa, että heille kaikille on määrättävä tehokasta kouristuslääkitystä.
Epileptisten kohtausten hoito suoritetaan vitamiinien ja kouristuslääkkeiden avulla. Angelmanin oireyhtymässä, johon liittyy kouristusoireyhtymä, B-vitamiinit sekä C-, D- ja E-vitamiinit ovat hyödyllisiä. Mutta vitamiinihoidon ottaminen itse on tässä tapauksessa erittäin vaarallista, koska hallitsematon vitamiinien saanti voi vähentää epilepsialääkkeiden tehoa ja aiheuttaa uusia, vakavampia ja pitkittyneempiä kohtauksia.
Myös kouristuslääkkeiden valinta ja niiden tehokkaan annostuksen määrääminen tulisi tehdä erikoislääkärin toimesta. Hän päättää myös, riittääkö yksi lääke vai tarvitseeko potilas kahta tai useampaa lääkettä pitkään.
Useimmille potilaille lääkärit määräävät valproiinihappolääkkeitä (Valproiinihappo, Depakine, Convulex, Valparin jne.), jotka estävät kohtauksia ja parantavat potilaiden mielialaa ja mielentilaa.
Valproiinihappoa on saatavilla tablettien, siirapin ja injektioliuosten muodossa. Suosituin lääke on pitkävaikutteinen lääke "Depakine" tabletteina ja laskimonsisäisenä liuoksena. Lääkäri määrää lääkkeen annostuksen yksilöllisesti potilaan painon, iän ja tilan perusteella.
Lääke otetaan aterioiden yhteydessä 2–3 kertaa päivässä. Keskimääräinen vuorokausiannos on 20–30 mg potilaan painokiloa kohden, enimmäisannos on 50 mg/kg päivässä.
Vasta-aiheet. Ei saa käyttää maksan ja haiman toimintahäiriöiden, verenvuototaipumuksen, hepatiitin, porfyrian tai lääkkeelle yliherkkyyden yhteydessä.
Sivuvaikutuksia ovat käsien vapina, ruoansulatus- ja ulostehäiriöt sekä painonmuutokset.
"Topiramaatti" on myös valittu lääke SA:lle. Sitä valmistetaan tablettimuodossa ja sitä käytetään sekä monoterapiana että yhdessä muiden lääkkeiden kanssa.
Antotapa ja annostus. Ota tabletit suun kautta ruokailusta riippumatta. Aloitusannos aikuisille on 25–50 mg vuorokaudessa, lapsille 0,5–1 mg/kg. Annosta suurennetaan viikoittain lääkärin ohjeiden mukaan.
Lääkettä ei tule käyttää raskauden ja imetyksen aikana, eikä myöskään yliherkkyyden sattuessa sen ainesosille. Lääkkeellä on monia erilaisia sivuvaikutuksia.
Lääkkeet, joita lääkäri voi määrätä Angelmanin oireyhtymään: Clomazepam, Rivotril, Lamotrigiini, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra jne.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Perinteinen lääketiede ja homeopatia
Perinteinen lääketiede, kuten homeopaattiset valmisteet, on tietenkin suhteellisen turvallista, mutta tällaisen hoidon tehokkuutta Angelmanin oireyhtymässä voidaan pitää kiistanalaisena.
Vaikka kansanhoito voi silti auttaa joissakin asioissa. Puhumme epileptisten kohtausten lopettamisesta. Tässä suhteessa yrttihoito voi olla varsin tehokasta.
Hyvän vaikutuksen antaa pioniin, lakritsiin ja limaskaan perustuva lääkekokoelma (komponentit otetaan yhtä suurina määrinä). Yrtit on jauhettava jauhoksi. Kahden viikon kuluttua ottamisen aloittamisesta kohtausten esiintymistiheys voi vähentyä merkittävästi.
Myös laventelinkeite (1 tl per lasillinen kiehuvaa vettä) on hyödyllinen kramppeihin. Seosta keitetään 5 minuuttia ja annetaan hautua puoli tuntia. Lääke otetaan yöllä 14 päivän ajan.
Äidinmarjan vesipitoista (tai alkoholipitoista) infuusiota pidetään tehokkaana epileptisten kohtausten hoidossa.
Angelmanin oireyhtymän kohtausten ehkäisyyn tarkoitetuista homeopaattisista valmisteista voidaan käyttää kamomillaan ja äitiyrttiin, Acidum hydrocyanicumiin, Argentum nitricumiin, Kalium bromatumiin ja Arsenicum albumiin perustuvia lääkkeitä. On kuitenkin otettava huomioon, että vain homeopaattinen lääkäri voi määrätä tehokkaita ja turvallisia lääkeannoksia kussakin yksittäistapauksessa.
Ennaltaehkäisy
Kuten lukija on luultavasti jo ymmärtänyt, lääketiede ei kuitenkaan vielä pysty estämään geenimutaatioita ja muita kromosomipoikkeavuuksia eikä korjaamaan tilannetta. Näin voi käydä kenelle tahansa, koska Angelmanin oireyhtymää sairastavat lapset syntyvät terveille vanhemmille, eikä genetiikka, joka on tällä hetkellä yksi vähiten tutkituista lääketieteen aloista, pysty vielä selittämään tätä.
Ainoa mitä voidaan tehdä, on suhtautua vastuullisesti raskauden suunnitteluun, ilmoittautua ja käydä tutkimuksissa ajoissa. Mutta jälleen kerran, tällainen toimenpide on enemmän opettavainen kuin ennaltaehkäisevä, kuten mitkään tutkimukset. Mutta nuoret vanhemmat tietävät etukäteen, mihin valmistautua, ja myönteisen vastauksen tapauksessa he päättävät, voivatko he ottaa vastuulleen sairaan lapsen kasvattamisen.
Ennuste
Angelmanin oireyhtymän ennuste riippuu kromosomipoikkeavuuden luonteesta ja sen havaitsemisen ajantasaisuudesta. Eniten kärsivät lapset, joiden kromosomissa 15 on "aukkoja" geeneissä (deleetio). Tällaisten potilaiden kävelyn ja puhumisen todennäköisyys on erittäin pieni. Muut tapaukset voidaan korjata huolellisella lähestymistavalla ja rakkaudella lasta kohtaan.
Valitettavasti tällaiset potilaat eivät pysty tulemaan yhteiskunnan täysivaltaisiksi jäseniksi, vaikka he eivät olekaan tyhmiä, vaan ymmärtävät puhetta ja sen merkitystä. Heillä on kuitenkin kommunikaatio-ongelmia koko loppuelämänsä ajan. Potilaille voidaan opettaa viittomakieltä lapsuudesta lähtien, mutta heitä ei voida pakottaa kommunikoimaan sanoin. "Puhuvien" potilaiden sanasto rajoittuu arkielämässä käytettyjen sanojen minimiin (5-15 sanaa).
Angelmanin oireyhtymää sairastavien potilaiden elinajanodotteen ja yleisen terveydentilan osalta luvut vaihtelevat keskiarvojen ympärillä. Aikuisuudessa potilaat kohtaavat enimmäkseen terveysongelmia, kuten skolioosia ja liikalihavuutta, jotka oikealla hoitomenetelmällä eivät ole hengenvaarallisia.